
Abstract
It is now possible to identify hereditary and acquired risk factors in a substantial percentage of patients presenting with a venous thrombotic event. The clinician is faced with an ever-growing number of laboratory tests that can be ordered in such patients, and there is considerable uncertainty as to how this information should be utilized in patient management. Some have argued that widespread testing of thrombosis patients for prothrombotic abnormalities such as the factor V Leiden and prothrombin G20210A mutations has been prematurely adopted into clinical practice as there are few data that their identification leads to improved clinical outcomes.
Dr. Rosendaal provides an overview of the epidemiology of venous thrombosis with an emphasis on hereditary and acquired risk factors. The presentation will include information obtained from properly designed case-control studies as well as family studies.
While some have suggested treatment strategies for managing patients with hereditary thrombophilia with prior thrombotic events or for managing patients undergoing procedures associated with increased thrombotic risk, clinical decision making is complicated by the need to assess the risk of recurrence and the likely benefit of prolonged anticoagulation versus the associated bleeding risk. Drs. Bauer, Heit, and Rosendaal discuss their approaches to patient management. Case presentations are used to illustrate the impact of laboratory test results on decisions.
I. The Epidemiology of Venous Thrombosis: Genetic and Acquired Causes
Frits R. Rosendaal, MD*
Klinische Epidemiologie en Hematologie, C9-P, Leids Universitair Medisch Centrum, Postbus 9600, NL-2300 RC Leiden, The Netherlands
Venous Thrombosis
The annual incidence of venous thrombosis is 1 to 3 individuals per 1000 per year.1,2 Its major manifestations are deep vein thrombosis (DVT) of the leg and pulmonary embolism. Thrombosis may also occur in other veins (upper extremities, liver, cerebral sinus, retina, mesenteric), which is rare. Major complications of venous thrombosis are a disabling postthrombotic syndrome, occurring in up to 20% of patients,3 and acute death from a pulmonary embolism, occurring in 1-2% of patients.2
The incidence of thrombosis is age dependent; it is extremely uncommon (1 in 100,000 per year) in childhood, and rises to nearly 1% per year in old age.4
Thrombosis
Obstructive clot formation in veins or arteries is the end product of an imbalance of procoagulant, anticoagulant, and fibrinolytic factors. Thrombosis may occur in arteries and veins. The most common manifestations of arterial thrombosis are myocardial infarction and ischemic stroke. In the mid-1800s, Virchow postulated three major causes of thrombosis: changes in the vessel wall, changes in the blood flow, and changes in the blood composition.5 These groups of causes do not have the same role in arterial and venous thrombosis, which explains why these two types of thromboses have overlapping but vastly different causes. In arterial thrombosis, vessel wall changes (atherosclerosis) and associated risk factors (hypertension, hyperlipidemia, smoking, diabetes mellitus) dominate. Because of the high blood pressure and blood flow in arteries, stasis and immobilization do not affect risk, and hypercoagulability of the blood has a relatively minor role. In venous thrombosis, on the other hand, stasis and immobilization are important risk factors, as are prothrombotic abnormalities. Venous thrombosis therefore has risk factors that are distinctly different from those for arterial disease; atherogenic factors such as smoking, hypertension, or hyperlipidemia do not increase the risk of venous thrombosis (Table 1 ).
Risk Factors for Venous Thrombosis
Environmental or acquired risk factors for thrombosis have been known for centuries and include immobilization, surgery, trauma, plaster casts, pregnancy, puerperium, lupus anticoagulants, malignancies, and female hormones.6,7 Familial thrombophilia was first described in the early 1900s, when pedigrees with an abundance of venous thrombotic events suggested a familial hypercoagulability. Genetic risk factors have been known since 1965, when Egeberg described the first family with an identified hereditary tendency to thrombosis, caused by antithrombin deficiency (previously known as antithrombin III).8 In the 1980s, protein C (PC) deficiency and protein S (PS) deficiency were described in familial thrombophilia.9,10 Major advances have been made during the past decade, with the discovery of several abnormalities in the clotting system that predispose to venous thrombosis. The relevance of these abnormalities lies in their high prevalence; contrary to the deficiencies of natural anticoagulants, they affect large numbers of people. Despite the availability of effective anticoagulant prophylaxis, acquired risk factors are still responsible for a large number of thrombotic events (Table 2 ).
Environmental or Acquired Causes of Thrombosis
Age
The risk factor with the greatest gradient of risk is undoubtedly age, with a 1000-fold difference in risk of thrombosis between the very young and very old.4 Thrombosis is not an exception in this, since virtually all diseases in humans are strongly dependent on age, either increasing or decreasing with age. As with other disorders, it is not clear why there is an age gradient. It is plausible that the reason is a combination of decreased mobility, decreased muscular tone, increased morbidity, and acquisition of other risk factors, as well as wear-and-tear on the veins themselves. The age effect is important, and it should always be taken into consideration in clinical decision making: a risk factor that increases risk 10-fold in the young, leads to an excess number of cases of only 9 per 10,000 (for a baseline risk of 1 in 10,000) in that age group, while the same relative risk of 10 would lead to additional hundreds of cases per 10,000 in the elderly. When weighing this risk against the side effects of treatment (e.g., prophylactic anticoagulation), the cost of screening (e.g., for prothrombotic mutations), or the disadvantages of not prescribing a treatment (e.g., female hormones), the decisions may well be different in different age groups. It is also important not to assume that risk estimates in one age group apply to another age group (i.e., we need to know from direct studies whether the relative and absolute risk in the young and the elderly are the same or different).
Malignancy
Venous thrombosis occurs often in cancer patients, as was first observed by Trousseau in 1865.11 Recurrent thrombophlebitis in various sites (saltans et migrans) is seen as a telltale sign for occult cancer, especially of the pancreas, and case reports have also linked DVT to cancer, both clinically overt and occult.
The thrombogenic effect of cancer may be the result of several factors, which probably all play a role to some extent. First, the tumor itself may directly increase the risk of thrombosis, due to humoral (production of procoagulants), mechanical (venous obstruction), and general (acute phase reactions) effects.12,13 Second, cancer may indirectly promote thrombus formation due to consequences of being ill (e.g., reduced mobility, reduced dietary intake of vitamins, such as folate). And third, there may be effects of treatment (e.g., surgical/radiologic scarring) or of chemotherapeutic treatment (e.g., tamoxifen in breast cancer,14 chemotherapy in testicular cancer15).
The prevalence of cancer among patients with venous thrombosis varies between 3% and 18% in studies. In a population-based Swedish study, 19% of patients with thrombosis had a malignancy known at the time of diagnosis, and cancer was diagnosed in the year after the thrombotic event in an additional 5%.16
Surgery and trauma
Without thromboprophylaxis, surgery will lead to thrombosis in up to 50% of the patients, dependent on the type of surgery.17,18 In orthopedic surgery of the hip and knee, the risk of thrombosis reaches 30% to 50%. The risk is also high in abdominal surgery (up to 30%), gynecologic surgery, and urologic surgery (in particular, radical prostatectomy).19 It is noteworthy that in orthopedic surgery, minor interventions such as arthroscopy lead to a considerable thrombotic risk.
Major trauma is also an important risk factor for thrombosis, which occurs in 50-60% of patients with head trauma, spinal injury, pelvic fracture, femoral fracture, and tibial fracture.20
Nowadays, these high risks are no longer observed because of the use of anticoagulant prophylaxis. Still, even in the Leiden region, with aggressive use of anticoagulants, we observed that 18% of patients with thrombosis had undergone surgery shortly before the thrombosis.21
Immobilization
Thrombosis may occur in many circumstances that are associated with immobilization, such as paralysis, bed rest, plaster casts, and prolonged travel.22–,24 All of these situations have in common the fact that they interfere with the function of the calf musculature in pumping the blood upstream though the veins. Even major traffic jams have been associated with venous thrombosis.25
A risk factor that has received renewed interest recently is air travel. The first cases of venous thrombosis after long-haul flights were reported in 1954.24 An interesting study on Heathrow Airport found that sudden deaths in the airport occurred far more often in the arrival hall than in the departure hall.26 Another recent study found a clear association between the duration of the flight and a 50-fold gradient of risk from flights under 5000 km to those over 10,000 km.27 Still, controlled studies have conflicting results on the magnitude of the risks (some even showing no risk increase at all), and it is unclear which factors contribute to risk.28–,31 In 2002, a large research project initiated by the World Health Organization (WHO) was launched to further elucidate these issues. The project is aptly named the WRIGHT study (WHO Research into Global Hazards of Travel). This initiative combines several studies into risks, mechanisms, and prevention of travel-related thrombosis, in which the roles of hypoxia and hypobaria, which have been shown to activate coagulation,32 will also be studied.
Oral contraceptives
Shortly after oral contraceptives were licensed in 1959, the first case of pulmonary embolism was reported in a nurse who started oral contraceptives because of endometriosis.33 Many reports have demonstrated that oral contraceptives, including the currently used low-dose oral contraceptives, increase the risk of thrombosis about 4-fold.34– 36
Most oral contraceptives contain an estrogen and a progestogen. The estrogen dose has over the years been reduced from 100 μg or more of ethinylestradiol to current formulations with 30 μg or less of ethinylestradiol. There is no convincing evidence that this has decreased the risk: both the earliest and the latest studies show 4- to 8-fold increases in risk for oral contraceptive users, and direct comparisons of oral contraceptives with 50 μg and 30 μg ethinylestradiol have yielded conflicting results.34,37 Progestogen content may also affect thrombotic risk: from 1995 onward, more than 10 studies have shown that oral contraceptives containing desogestrel and gestodene (so-called third-generation progestogens) confer a 2-fold higher risk of venous thrombosis than formulations containing levonorgestrel (so-called second-generation progestogen).34,38– 40
Hormonal replacement therapy
After it was shown that hormone replacement with only estrogens increased the risk of endometrial cancer, it became common to combine an estrogen with a progestin, except in women without a uterus. The estrogens in most preparations are conjugated estrogens derived from pregnant mare urine, while the progestin usually is medroxyprogesterone.41 In addition to oral administration, transdermal patches and subcutaneous pellets are available. It has recently become clear from several studies that hormonal replacement therapy (HRT) is also associated with a 2- to 4-fold increased risk of thrombosis.42– 44
Pregnancy and puerperium
About 1 in 2000 women will develop thrombosis during pregnancy. This risk is about 10-fold that of the risk for nonpregnant women of the same age.45,46 The risk is also increased in puerperium, with most studies finding a higher risk than during pregnancy. Anticoagulant prophylaxis is therefore more frequently prescribed postpartum than during pregnancy; other factors contributing to this practice include the long antepartum period of treatment with heparin injections and the option of using oral anticoagulation post-partum.
Antiphospholipid antibodies
The risk of thrombosis is increased in patients with antiphospholipid antibodies, both among those with systemic lupus erythematosus (SLE) (half of whom have these antibodies) and among those with isolated antiphospholipid antibodies. The clinical presentation and the risk of thrombosis vary widely between patients. Overall, the risk is about 10-fold increased.47– 49
Genetic Causes of Thrombosis
Deficiencies of natural coagulation inhibitors
Deficiencies of antithrombin, protein C, and its cofactor protein S were the first genetic causes of venous thrombosis to be discovered. All 3 deficiencies are rare and are found in less than 1% of the population (type I antithrombin deficiency in less than 1 per 1000).50–,52 Even among patients with thrombosis, only a small percentage carry one of these defects.53,54 Because these abnormalities are so rare, population-based studies to estimate risk are scarce, and most information has been gathered in family studies. Because thrombophilic families may harbor more than one genetic defect, including unknown defects that may epistatically interact with the deficiency of a coagulation inhibitor, it is unclear whether the risk estimates for carriers of the defects in these families can be extrapolated to other individuals with these abnormalities.55 Generally, these deficiencies appear to increase risk about 10-fold in heterozygotes.54,56,57 Most clinicians feel that antithrombin deficiency carries a higher risk than PC or PS deficiency. The exceedingly rare homozygous deficiency of a natural anticoagulant results in a pronounced thrombotic tendency with widespread thrombosis (purpura fulminans) occurring shortly after birth.58,59
Factor V Leiden
Among Caucasians, factor V Leiden (factor V R506Q) is the most common genetic defect predisposing to thrombosis, with an overall prevalence of carriers of around 5%.60,61 However, because of founder effects, the population prevalence may vary widely between different regions; for example, in southern Sweden it is 15%.61 Factor V Leiden is found in 20% of unselected patients and in up to 50% of selected patients with venous thrombosis (e.g., positive family history in first degree relatives, first event prior to age 50).62
Factor V Leiden causes thrombosis via the intermediate phenotype of resistance to activated PC (APC resistance).63 This intermediate phenotype is the result of a mutation at one of the cleavage sites in factor V, where APC inactivates factor Va.60 Factor V Leiden increases the risk of venous thrombosis 3- to 8-fold in heterozygous carriers, and 50- to 80-fold in homozygous carriers.62,64 Homozygosity, which is not exceedingly rare due to the high allelic prevalence, is found in about 1 per 5000 persons in the population.
Prothrombin 20210A
This mutation in the 3′-untranslated region of prothrombin at position 20210 (G to A, PT20210A) is found in about 3% of Caucasians, again with regional variations in prevalence ranging from 1% to 6%.65,66 Among patients with venous thrombosis enrolled in the Leiden Thrombophilia Study, this mutation is present in 6%.65 It increases the risk of thrombosis about 3-fold, which appears to be mediated through elevated prothrombin levels.65
Blood group
ABO blood group is associated with the risk of venous thrombosis, with an increased venous thrombotic risk in those with non-O blood groups (2- to 4-fold increased risk).67 These individuals also have higher von Willebrand factor levels and higher factor VIII levels, and it is likely that blood group is related to thrombotic risk via its relation to von Willebrand factor and factor VIII levels.68
Methylene tetrahydrofolate reductase 677T
A variant in the gene for methylene tetrahydrofolate reductase (MTHFR), which plays a role in homocysteine metabolism, has been shown to be associated with mildly elevated homocysteine levels.69,70 The variant is common (about 10% of the general population are homozygous carriers), but the elevation of homocysteine levels is so small that little effect on risk can be expected.71 It has been estimated that if hyperhomocysteinemia is a true cause of thrombosis, homozygous carriers of the MTHFR 677T variant have no more than a 16% increased risk of venous thrombosis.
Other Plasma Abnormalities Associated with the Risk of Thrombosis
Hyperhomocysteinemia
Homozygous deficiency of cystathionine beta-synthase (CS) causes high plasma levels of homocystine, presence of homocystine in urine (hence the name homocystinuria), and atherosclerosis, arterial disease, and venous thrombosis occurring at a young age.72 More recently it has become clear that mildly elevated levels of homocystine (over 18 μmol/L) are also associated with an increased risk of thrombosis.73– 75 Such levels are found in 5-10% of the general population and roughly double the risk of venous thrombosis. Hyperhomocystinemia is usually the result of acquired causes (low intake of folate or vitamins B6 or B12) and only rarely of heterozygous CS deficiency.
High levels of clotting factors
Elevated levels of prothrombin (factor II), factor VIII, factor IX, or factor XI, as well as thrombin activatable fibrinolysis inhibitor (TAFI), are all associated with an increased risk of thrombosis.65,68,76–,78 When a cut-off level is set at the 90th percentile of the distribution in the general population, the risk of thrombosis is 2- to 3-fold increased in individuals exceeding that level, which by definition is present in 10% of the population. Although little is known about the origins of such elevated levels, it is likely that these are a combination of genetic and acquired causes. For high levels of factor VIII, it has been shown that there is familial clustering of high levels, even when the blood group effect has been taken into account.79,80
Thrombosis as a Multicausal Disease
Many risk factors for thrombosis known today are common in the general population, and therefore several will often be present in one individual. Acquired risk factors also have a high prevalence, and therefore a combination of several genetic and several acquired risk factors in one individual may not be infrequent. It is likely that the combined presence of several of these factors, and their interactions, are required for thrombosis to develop.
Gene-Gene and Gene-Environment Interaction
High risks of thrombosis have been seen for combinations of natural coagulation inhibitors (PC, PS, antithrombin) deficiency and factor V Leiden or prothrombin 20210A.81– 85 The high risk of thrombosis in homozygous defects can also be seen as an example of gene-gene interaction.
In families with thrombophilia caused by deficiencies of PC, PS, and antithrombin, the risk of thrombosis is considerably increased during pregnancy, puerperium, or use of oral contraceptives.86–,88 In population-based studies, a synergistic effect (gene-environment interaction) has been shown for factor V Leiden and use of female hormones, either as oral contraceptives or as postmenopausal hormone replacement,89,90 as is shown in Figure 1 .
Conclusions
The occurrence of venous thrombosis is the result of interacting genetic and acquired causes. More knowledge of risk factors, particularly of the interactions of risk factors, will enable us to individualize risk, and in that way reach individualized guidelines for thrombosis prevention.
II. Case Presentations: What Is the Clinician to Do?
Kenneth A. Bauer, MD,*
VA Medical Center, 1400 VFW Parkway, West Roxbury, MA 02132
Mayo Clinic, Plummer 549, 200 First St. SW, Rochester, MN 55905
Before the case presentations were made, the following question was posed to the three speakers: “In the clinical evaluation of patients referred to me (or our center) with venous thromboembolic disease, which of the following laboratory tests are obtained?” The responses are listed in Table 3 and attention should be paid to the caveats in the footnote. While there are differences in our practices, there is general agreement on the analytes that should be measured as part of the “hypercoagulable workup.”
These actual cases (of Dr. Bauer) were selected for discussion as they illustrate more “nuanced” issues related to the diagnosis or long-term management of hereditary thrombophilia. Most of the cases involve the less commonly encountered hereditary thrombophilias for which there are little data upon which to base patient management decisions.1 While the risk of incident and recurrent venous thromboembolism or complications of pregnancy is increased in association with deficiencies of antithrombin, PC, or PS, the magnitude of those risks is uncertain. Estimating the most appropriate duration of oral anticoagulation therapy is especially difficult and requires an assessment of the rates of venous thromboembolism recurrence (including fatal pulmonary embolism) and major bleeding (including fatal bleeding) over time for the individual patient. Randomized clinical trials addressing duration of anticoagulation have included too few patients with these deficiencies to draw firm conclusions.2–,5 Patient compliance has a major impact on the success of therapy, and patient preferences must be factored into this decision. In this circumstance, decision analysis can be helpful, especially sensitivity analyses, which show the impact of varying rates of recurrence or bleeding over a plausible range, on the decision to treat or withhold therapy.6 The speakers will therefore have different opinions regarding management of some of the cases.
What data should we use to estimate the risk of recurrent venous thrombosis?
Dr. Heit:
In population-based epidemiologic studies from Olmsted County, Minnesota, 23.5% of patients with acute pulmonary embolism presented as essentially sudden death (i.e., survived less than one day, or pulmonary embolism was discovered at autopsy and judged to be the cause of death).7 Seven-day mortality after acute pulmonary embolism was 28.9%.7 I believe these rates are higher than those usually quoted because the Olmsted County autopsy rate was almost 70% and most of these events were discovered at autopsy. Among 1719 Olmsted County residents with incident venous thromboembolism who survived at least one day, 456 developed recurrent venous thromboembolism over 16,430 person-years of follow-up (Heit, unpublished observations). The overall 7-day case-fatality associated with recurrence was 16.7% (30.7% for recurrent pulmonary embolism and 4.2% for acute DVT). Many of the recommendations I make regarding therapy reflect these rates. Thus, if one uses the overall rate for recurrent venous thromboembolism case-fatality, prolonged anticoagulation would be favored for almost all ages.8
Dr. Bauer:
Standard therapy for patients with DVT and pulmonary embolism typically includes anticoagulation for 3-6 months with warfarin at an International Normalized Ratio (INR) between 2 and 3. Several randomized clinical trials have provided data that the incidence of recurrent venous thromboembolism following the cessation of therapy in patients presenting with a first episode of unprovoked symptomatic venous thromboembolism is 5-15% in the first year and 20-30% at 4 years.2–,5 However, estimates of the risk of fatal pulmonary embolism in patients following adequate treatment for an initial episode of symptomatic DVT or pulmonary embolism are very low at ~0.4% per year.9 Recurrences occur much less frequently when the initial event is associated with transient risk factors.
As noted above, we have little information on the risk of recurrent thrombosis in unselected patients with deficiencies of antithrombin, PC, or PS; most reports in the literature report selected kindreds with strong clinical penetrance and high recurrence rates. Among patients with a first episode of venous thromboembolism associated with the more common factor V Leiden or prothrombin G20210A mutations, the data disagree on whether their recurrence risk is higher compared to those without a prothrombotic mutation;4,10–,16 the current consensus view is that the presence of either of these abnormalities should not influence the duration of anticoagulant therapy. However, there does appear to be a significantly higher recurrence risk for the small subset of patients who are heterozygous for both mutations.17 While prolonged anticoagulation at an INR of 2 to 3 is highly effective in preventing thrombotic recurrences, this benefit is partially offset by major bleeding, which occurs at a rate of 2-3% per year.3 While this annual risk is likely lower among young, compliant patients without risk factors for bleeding on warfarin therapy,18 the risk of bleeding over a lifetime is substantial. Thus the patients who will benefit from long-term anticoagulant prophylaxis at an INR of 2-3 will be mainly those with multiple idiopathic venous thrombotic events in close temporal proximity.
Case #1
Type 1 antithrombin deficiency: Lifelong anticoagulation for a 17-year-old after a first event?
A 17-year-old male without significant past medical history sustained symptomatic DVT of the left leg in December 2000. Ultrasound showed thrombosis involving the popliteal and common femoral veins as well as the greater saphenous vein. He was hospitalized and treated with intravenous heparin and warfarin. He responded well to treatment, and the pain and swelling in the left leg resolved after 1 month. There was no antecedent history of trauma or illness prior to the DVT. Laboratory evaluation on admission: Hgb 12.2, Hct 35, Plts 457K, prothrombin time (PT) 15.3 secs, partial thromboplastin time (PTT) 28.6 secs; cardiolipin antibody levels, Staclot LA, and dilute Russell viper venom time within normal limits; homocysteine 6.1 mmol/L; and factor V Leiden and prothrombin 20210A mutations not present. Antithrombin, PC, and PS measurements were not done initially.
The patient remained on warfarin at an INR of 2-3 for 6 months and tolerated the medication well without any evidence of bleeding. Since the initial DVT, he noted occasional swelling in his left leg and purplish discoloration of the left foot with increased vascular markings. Repeat ultrasound in November 2001 showed changes in the left leg, which were read as consistent with old thrombophlebitis. Further laboratory evaluation was performed several months after the cessation of warfarin in December 2001: PT 12.7 secs, PTT 26.9 secs, antithrombin functional activity 53% (normal range 75-135%), normal PC antigen and activity, PS functional activity 93%, total PS antigen 76%, and free PS antigen 126%. Repeat antithrombin activity in January 2002 was 54% with an antithrombin antigen level of 57%.
The patient is told he has type I antithrombin deficiency and comes to you for a second opinion regarding whether he needs to restart anticoagulation. He has fully returned to his normal activities, although he has some residual swelling and discoloration of his left leg. He has never undergone any surgical procedures and does not take any medications. There is no definite history of venous thromboembolism in his family, though there is a question as to whether his maternal grandfather developed phlebitis in his 40s. His mother had two uncomplicated pregnancies, but another was complicated by preeclampsia. He is an active high school student and does not smoke, drink, or use illicit drugs.
Rationale for long-term anticoagulation (Dr. Heit):
For this patient, I would recommend indefinite oral anticoagulation therapy targeting an INR of 2.5 (therapeutic range 2.0-3.0) as the patient had an unprovoked proximal DVT at a very early age in the presence of antithrombin deficiency. The risk of recurrent venous thromboembolism would significantly outweigh the risk of anticoagulant-related bleeding.8,19,20 This recommendation assumes that the patient has no risk factors for anticoagulant-related bleeding and is reliable in having his INR monitored on a routine basis. In support of this recommendation, the patient tolerated his previous course of anticoagulation well without any evidence of bleeding. I would recommend that all symptomatic family members (i.e., particularly the patient’s mother and grandfather) be tested for plasma antithrombin activity to determine carriership for antithrombin deficiency. All asymptomatic adult female family members at risk for having antithrombin deficiency and considering oral contraception, pregnancy, or estrogen replacement therapy also should be tested. Testing all asymptomatic adult family members is controversial, because male family members should receive prophylaxis if exposed to a high-risk situation for venous thromboembolism (e.g., major surgery), regardless of whether or not they are carriers of antithrombin deficiency. Furthermore, it is uncertain whether asymptomatic family members with antithrombin deficiency should receive a higher intensity or longer duration of prophylaxis. I would not recommend chronic anticoagulation for asymptomatic antithrombin-deficient family members.
An alternate viewpoint (Drs. Bauer and Rosendaal):
The penetrance of venous thrombosis in this family, some of whom are certain to be antithrombin deficient, appears low. While isolated type I antithrombin deficiency is the hereditary thrombophilia considered to have the highest clinical penetrance within selected families, we really do not have much information about the true risk of recurrence in this type of patient. Lifelong anticoagulation after a single DVT in a young active individual has major potential ramifications on his lifestyle and psyche. The pros and cons of indefinite anticoagulation should be thoroughly discussed with the patient and his family and their preference taken into account. The risk of fatal spontaneous pulmonary emboli without premonitory symptoms should be very low in this patient and does not justify long-term anticoagulation per se. On the other hand, oral anticoagulation perhaps carries an average annual risk of 1-2% of major hemorrhage requiring hospital admission and 0.5% of intracranial hemorrhage. While these risks may be substantially lower among young people, they will become large when viewed over this patient’s anticipated life expectancy. We recommend no long-term anticoagulation, a compression stocking for the left lower extremity, and advice regarding prophylaxis in high-risk situations.
Case #2
Air travel and thrombophilia: To prophylax or not to prophylax?
A lean 19-year-old female college student with confirmed type I antithrombin deficiency is referred for consultation. She was identified as being antithrombin deficient 4 years ago when oral contraceptive use was being considered for the purpose of regulating her irregular and painful menses. It was known that her maternal twin uncles both have antithrombin deficiency and first sustained venous thromboembolism following football injuries while in college; they have had several pulmonary emboli since then despite being maintained on warfarin. The patient’s mother was discovered to be antithrombin deficient while pregnant and was prophylaxed with heparin during the pregnancy. She has never had a venous thromboembolic event to date or miscarriages and is not maintained on anticoagulants. The patient is an only child.
Other than a history of supraventricular tachycardia starting 3 years ago, the patient is healthy and has never been pregnant or on oral contraceptives. She does not smoke or drink alcohol. Physical examination is unremarkable. Her antithrombin activity level is 45% of normal, and the remainder of her hypercoagulable workup is negative. The patient is planning a summer trip to Asia and will be on a plane for approximately 20 hours; her mother is very anxious regarding the risk of venous thromboembolism associated with prolonged travel. Would you recommend pharmacological antithrombotic prophylaxis when she travels to Russia?
An individualized decision (Drs. Bauers and Rosendaal):
There is evidence that prolonged air travel (> 3000 miles) is a risk factor for venous thromboembolism, and this risk is likely increased significantly in patients with hereditary thrombophilia. The absolute risk in this patient, however, remains small. It was recommended to the patient that she try to ambulate and exercise her legs as much as feasible in flight. Graduated compression stocking therapy (calf-high, 20-30 mm of mercury graduated compression stockings) during the long-haul plane flight could also be prescribed. Whether more aggressive prophylaxis is warranted is unknown. However, a single subcutaneous injection of a prophylaxis dose of a low molecular weight heparin (e.g., dalteparin sodium 2500-5000 IU or enoxaparin sodium 40 mg) immediately prior to boarding the airplane would be reasonable. After discussing the various approaches with the patient, she was prescribed low molecular weight heparin primarily to allay the mother’s anxiety. The presence of antithrombin deficiency should not diminish the efficacy of low molecular weight heparin in preventing thrombosis. Following her return from her trip, the patient was seen and reported that she never took the low molecular weight heparin and did not suffer a thrombotic event.
Case #3
A) Difficulty of making a diagnosis of PS deficiency even in the backdrop of a thrombophilic family said to have the disorder.
A 33-year-old white male who sustained a proximal right leg DVT 6 weeks ago is referred for evaluation. He was treated with unfractionated heparin intravenously in the hospital and is currently on warfarin. The patient has no prior history of thrombosis or other medical illness. Family history is positive for recurrent venous thrombosis in his paternal uncle starting at age 15; he is now maintained on warfarin and reportedly has PS deficiency. His daughter is also deficient, but has not yet had a thrombotic event. According to the uncle, several other female relatives have had phlebitis. The patient’s father never had venous thromboembolism but has hyperlipidemia and sustained a myocardial infarction 3 years ago. The patient’s mother and 2 sisters have not yet sustained venous thrombosis or miscarriages. Physical examination is remarkable for mild swelling of the distal right lower extremity.
Laboratory data on admission: complete blood count (CBC), PT, and PTT within normal limits; functional PS level 57% (normal range 70-140%) and total PS antigen 54%; APC resistance screen 2.7 (normal > 2.0); antithrombin and PC levels within normal limits; prothrombin 20210A mutation not present; factor VIII:C 63%; cardiolipin antibody levels within normal limits; fasting homocysteine level 11.5 mmol/L (reference range reported as < 8.9). A test for a lupus anticoagulant (dilute Russell viper venom time) performed 3 weeks later, while the patient was stably anticoagulated on warfarin, was positive.
Laboratory records regarding the diagnosis of PS deficiency in the patient’s uncle and his uncle’s daughter are not available. Given that a diagnosis of type I PS deficiency is a likely possibility in this patient, you obtain a total PS antigen level of 44% at a time when the patient is stably anticoagulated with an INR of 2.7.
Does the patient have hereditary PS deficiency?
Discussion (Drs. Heit and Bauer):
The patient is a 33-year-old man with unprovoked proximal right leg DVT and a presumed family history of symptomatic PS deficiency. At the time of admission (and presumably prior to beginning anticoagulation therapy), the PS activity was reduced to 57%. Interpretation of PS levels during an acute illness can be difficult. About 60% of plasma PS circulates bound to a complement regulatory protein (C4b-binding protein). Only unbound (“free”) PS acts a cofactor for activated PC-mediated inactivation of procoagulant factors Va and VIIIa. Because C4b-binding protein plasma levels increase during acute illness, the percentage of bound plasma S also increases with a proportional reduction in free plasma S (e.g., a type III PS deficiency pattern). Whether an acquired type III PS pattern predisposes to thrombosis remains controversial. However, in this patient we were told that the total PS antigen also was reduced, suggesting a true congenital (Type I) PS deficiency. While measurement of free PS antigen level might have been helpful in further corroborating this impression, we would not diagnose this patient as having congenital PS deficiency based on measurements made during an acute illness. Although we are told that the patient’s “APC resistance assay” was normal, we are not told what “generation” APC resistance assay was performed. If the APC resistance assay was a “second generation” assay (i.e., performed after premixing the patient’s plasma with factor V deficient plasma), then factor V Leiden can be excluded with confidence. However, “first generation” APC resistance assays are relatively insensitive. In the Mayo Clinic laboratory, heterozygous factor V Leiden carriers may have APC-resistance ratios as high as 2.9 when a first-generation APC resistance assay is used. The factor V Leiden carrier status for this patient is important because carriers tested with an earlier generation PS activity assay often had a falsely reduced PS activity. Later generation functional activity assays partially “corrected” for this problem by adding bovine factor V. Thus, if a first-generation APC-resistance assay was performed, I would retest with a second-generation APC-resistance assay or perform a direct DNA-based assay for the factor V Leiden mutation. The total PS antigen level of 44% while the patient was stably anticoagulated on warfarin was not helpful; a result of 25% or less would have suggested that the patient was a carrier of type I PS deficiency. Functional protein S assays cannot be interpreted in patients on oral anticoagulants. The positive tests for a lupus anticoagulant were false-positives due to oral anticoagulation, which was verified subsequently.
After 6 months of anticoagulant therapy, warfarin is discontinued and the patient undergoes further laboratory evaluation 2 weeks later. His PT is 13.7 sec with PTT of 33.2 secs; thrombin time is normal at 11.2 secs. Functional PS level is 38% (normal range 70-140%) and free PS is 40% (normal range 46-122%) with a total PS antigen of 90% (normal range 77-124%); factor VIII activity is now 160%. Tests for a lupus anticoagulant and an APC resistance assay using factor V-deficient plasma give normal results. A follow-up ultrasound shows recanalized clot in the common femoral, superficial femoral, and popliteal veins. One month following discontinuation of warfarin, PS activity was remeasured at 48% with free and total PS antigen of 47% and 91%, respectively. Clinically, the patient is doing well with some residual right leg swelling. His occupation requires him to spend a large part of his day on his feet, and he chronically wears a thigh-length compression stocking.
Discussion (Drs. Heit and Bauer):
Repeat testing performed 2 weeks after stopping warfarin therapy showed persistent reduction in plasma PS activity and free PS antigen. However, the total PS antigen is now normal, leading to the suspicion that the patient has a type III PS deficiency; this result, however, is at variance with results obtained at the time the patient presented with an acute DVT when all three PS measurements were reduced. Type I and type III PS deficiency can be found in individuals from the same family as verified by DNA mutation analysis. We recommend repeat PS testing yet a third time in several weeks, which supported the diagnosis of type III PS deficiency. Given a history of an unprovoked DVT, his family history, and his personal preference, a decision was made to chronically anticoagulate the patient with a target INR of 2-3. It is not necessary to use heparin or low molecular weight heparin before restarting warfarin. Interestingly, the factor VIII:C measurement that was obtained acutely in the setting of the acute DVT was only 63%, while the level obtained following the cessation of oral anticoagulation was somewhat elevated at 160%; these results did not influence our decision regarding long-term anticoagulation, and we are not generally obtaining factor VIII:C levels in such patients until more data become available with regard to the relationship between elevated levels and risk of recurrence.
Another viewpoint (Dr. Rosendaal):
It should be emphasized that there are few data indicating that the recurrence risk is higher in PS deficiency than in patients with unprovoked DVT without hereditary thrombophilia. Age is also a risk factor for hemorrhage during anticoagulation, so it is unclear whether the benefits of indefinite anticoagulation at an INR of 2-3 would outweigh the risks. Therefore, I would not advocate prolonged treatment with anticoagulants, and this policy would not be influenced by the absence or presence of PS deficiency.
B) Management decisions surrounding pregnancy in the patient’s sisters.
When you initially see the patient, he tells you that his 38-year-old sister is early in the second trimester of a pregnancy. Her past medical history is remarkable only for hypothyroidism. She had previously used oral contraceptives without complications and is G3 P0 Ab 2 (therapeutic). She does not smoke. No hypercoagulable evaluation is undertaken. You subsequently learn that she was hospitalized at 38 weeks’ gestation for preeclampsia for several days and then underwent a cesarean section. She was managed with pneumatic compression boots postpartum but at 4 weeks postpartum was diagnosed as having pulmonary emboli and a proximal right leg DVT. She was admitted to the hospital and treated with heparin followed by warfarin. After she recovers, you see her in consultation. She has not yet been evaluated for PS deficiency, and you elect to wait to evaluate her until she has completed 6 months of anticoagulation.
Two months later, the patient’s 41-year-old sister, who is now 30 weeks pregnant, sees you in consultation regarding anticoagulation during pregnancy. She is G5 P1 Ab 2 with 1 miscarriage at 7 weeks. She has not had any thrombotic events and her child was delivered vaginally 2½ years ago following an uncomplicated pregnancy. She did not receive prophylactic anticoagulation. She also had used oral contraceptives while in her 20s without complications. A functional PS level was 62% of normal at 2 months’ gestation.
Discussion (Drs. Heit, Bauer, Rosendaal):
With regard to the patient’s 38-year-old sister, we would not recommend indefinite anticoagulation whether or not she is proven to have PS deficiency. In community-based epidemiologic studies performed at the Mayo Clinic, pregnancy- or postpartum-associated venous thromboembolism was associated with a lower risk for recurrent venous thromboembolism than venous thromboembolism in the absence of pregnancy.21,22 Additional provocative factors that further increased her risk of peripartum venous thromboembolism include preeclampsia and cesarean section. Several weeks following the completion of oral anticoagulation, her functional PS, free PS antigen, and total PS antigen levels were 20% (normal range 51-133%), 40% (normal range 62-146%), and 86% (normal range 70-140%), respectively, confirming the diagnosis of type III PS deficiency.
With regard to the patient’s pregnant 41-year-old sister, the diagnosis of hereditary PS deficiency is uncertain because pregnancy causes reduced PS levels. The incidence of venous thromboembolism is higher during the postpartum period than during pregnancy. Therefore, for this patient, I would recommend only prophylaxis for 4-6 weeks postpartum. Either low molecular weight heparin at prophylactic doses (dalteparin sodium 2500 IU SC qd or enoxaparin sodium 40 mg SC qd) or unfractionated heparin followed by warfarin with a target INR of 2-3 could be used.
We also recommend testing of the patient’s father to provide further support for a diagnosis of hereditary PS deficiency. A report is subsequently received from his physician that his functional PS level is low at 38% (normal range 70-150%). He has not yet had a venous thromboembolic event though he has had a myocardial infarction.
Case #4
Homozygosity for factor V Leiden: should one anticoagulate a 36-year-old woman after a major episode of superficial venous thrombophlebitis in the setting of oral contraceptive use?
A 36-year-old woman on a third-generation oral contraceptive developed pain and erythema on the medial aspect of her right thigh in October 2000. Ultrasound evaluation on October 13, 2000, showed superficial venous thrombophlebitis with involvement of the greater saphenous vein, which did not extend to the saphenofemoral junction (clot was 10 cm away from it) or involve the deep veins. The patient was advised to take aspirin or a nonsteroidal antinflammatory drug, but the pain progressed and a repeat ultrasound on October 19 showed the thrombus was now 24 cm in length but again did not extend to the saphenofemoral junction or involve the deep veins. Laboratory evaluation was performed and the results, which returned one week later, demonstrated homozygosity for the factor V Leiden mutation. This prompted performance of yet another ultrasound that showed that the greater saphenous vein thrombus was now 21 cm and the clot was stated to be poorly adherent and 8 cm from the saphenofemoral junction.
She had been on oral contraceptives from 1992 to 1996 and then had 2 uncomplicated pregnancies. She has not had any miscarriages and resumed the pill 11/2 years ago. She is on no other medications.
She was admitted to the hospital and begun on therapeutic doses of low molecular weight heparin and warfarin. Repeat ultrasound on October 30 showed that the thrombus extended to within 1 cm of the saphenofemoral junction. She received low molecular weight heparin for 4-6 weeks and was treated with warfarin with a target INR of 2-3 for 6 months. Follow-up ultrasound on November 7 showed that the thrombus had regressed and was now 8 cm from the saphenofemoral junction. The patient has done well since then and has been maintained on warfarin with good control of her INR.
Many members of her family, including her single female sibling, have already been screened and found to be heterozygous for factor V Leiden. There is no history of thrombosis on the maternal side of the patient’s family save for her maternal grandmother who was said to have “varicose veins and swollen legs”; her factor V Leiden status is unknown. The patient’s father had an episode of undiagnosed hemoptysis a number of years ago. He is heterozygous for factor V Leiden and has 10 siblings, one of whom had a pulmonary embolism and DVT and is heterozygous. A paternal cousin of the patient died of pulmonary embolism at age 47 late in the year 2000. Another paternal cousin has a history of cerebral thrombosis and another had a cerebral aneurysm.
After 6 months of anticoagulation, warfarin was discontinued and further laboratory evaluation was undertaken. Laboratory evaluation: CBC was normal; levels of antithrombin, PC, PS, and factor VIII:C were normal; lupus anticoagulant testing was negative; and cardiolipin antibody levels were in the normal range. Physical examination is completely normal.
Discussion (Drs. Heit and Rosendaal):
At the least, we would recommend an alternative form of contraception to this patient’s third-generation oral contraceptives (containing the progestin desogestrel or gestodene). One could consider switching to a second-generation oral contraceptive (containing the progestin levonorgestrel), since the risk of venous thromboembolism appears to be less compared with first- or third-generation oral contraceptives. This would be a reasonable recommendation if the patient were to remain on indefinite anticoagulation therapy. If indefinite anticoagulation is recommended, a case can be made that the most reliable form of contraception should be used (i.e., mandatory use of oral contraceptives assuming tubal ligation/vasectomy is not an option) in view of the teratogenic effects of coumarin derivatives. An alternative would be to switch to a nonhormonal form of contraception and stop oral anticoagulation after some period of therapy (e.g., 3 months is adequate). We would favor the latter recommendation, because the risk of venous thromboembolism recurrence is significantly less among women with oral contraceptive-associated venous thromboembolism than for women with venous thromboembolism not associated with oral contraceptives.21,22 While the risk of venous thromboembolism recurrence is increased among homozygous factor V Leiden carriers, it is unclear whether this risk exceeds the risk of chronic anticoagulant-associated bleeding.
Consultant’s recommendation (Dr. Bauer):
This patient does not meet any of the usual criteria for chronic anticoagulation (i.e., superficial venous thrombosis in the setting of oral contraceptive use). She was comfortable and happy with nonhormonal methods of contraception and did not plan further pregnancies. Given that she is homozygous for factor V Leiden mutation, her risk of sustaining an unprovoked major venous thromboembolic event is estimated to be over 50% during the remainder of her lifetime with a risk of ~0.5-1% per year.23 She has no risk factors that place her at increased risk for anticoagulant-induced bleeding. The patient had major concerns regarding morbidity, particularly postphlebitic syndrome, should she sustain a DVT. Following a long discussion of the risks of thrombosis with anticoagulation vis-à-vis bleeding without anticoagulation, she had a clear preference for long-term anticoagulation with a target INR of 2-2.5. This was in agreement with the opinion of the hematologist who had seen her previously; indeed the patient was so motivated that she had a home INR monitoring instrument in order to closely monitor her anticoagulation intensity.
Case #5:
Two episodes of unprovoked venous thromboembolism (VTE) separated by 12 years: Do you anticoagulate indefinitely?
An obese 54-year-old male sustained pulmonary emboli documented by a high probability ventilation-perfusion lung scan after presenting with gradually increasing shortness of breath during the preceding 3 months, which had been attributed to being overweight and out of shape. He also noted some pressure in his left lower extremity 3 weeks before presentation. He was hospitalized and anticoagulated with heparin followed by warfarin. Laboratory evaluation prior to the initiation of warfarin showed a slightly low antithrombin level of 72% with normal PC and PS levels. Plasma homocysteine was normal and the IgG cardiolipin antibody level was at the high end of the normal range at 15 GPL U/mL.
Past medical history is remarkable for an unprovoked DVT in the left leg 12 years ago for which he was hospitalized and treated with 6 months of anticoagulation. To his usual weight of 195 lbs, he has added 80 lbs in the past 3 years. Family history is positive for blood clots in his father, in his 70s, following a fall at a baseball game; he died of a myocardial infarction at age 82. The patient has 2 children who are alive and well. He works as a school superintendent, does not currently smoke, and drinks alcohol only socially.
Physical exam is unremarkable save for 1+ edema in the left leg. Further laboratory evaluation: heterozygosity for the factor V Leiden mutation, the prothrombin G20210A mutation was not present; repeat plasma antithrombin activity level was 87% of normal; lupus anticoagulant was not present; IgG cardiolipin antibody level was minimally elevated at 17.7 GPL U/mL (normal 0-15); and factor VIII coagulant activity was elevated at 170% of normal (normal range 50-150%).
He has now completed 6 months of anticoagulant therapy and is referred for consultation regarding continued anticoagulation.
Discussion (Dr. Heit):
I would recommend indefinite anticoagulation because obesity and older age are both independent risk factors for venous thromboembolism recurrence21,22 and because the patient’s most recent episode was unprovoked. The contribution of the combination of an increased factor VIII:C, a weakly positive IgG isotype anticardiolipin antibody, and 1 factor V Leiden allele to the risk of venous thromboembolism recurrence is uncertain. Some studies have shown an increased risk of recurrence among heterozygous factor V Leiden carriers, but most have not. One study has reported that elevated factor VIII:C levels are associated with a significantly increased recurrence risk;24 this data will not, however, influence my management decision until corroborating data are available. Most studies have suggested that the risk of thrombosis associated with IgG isotype anticardiolipin antibodies is increased at levels of 30-40 GPL U/mL, but the implications of persistent low-level elevations of IgG cardiolipin antibody are uncertain.
Discussion (Dr. Rosendaal):
There is little information that the laboratory results on this patient or his obesity place him at a higher risk than others for recurrent venous thrombosis; thus it is unclear whether the benefits of long-term anticoagulation outweigh the risks. I would therefore not place this patient on prolonged anticoagulation, unless he clearly wishes it.
Consultant’s recommendation (Dr. Bauer):
While this patient is definitely at risk for recurrent venous thromboembolism (~10% risk in the first year after 6 months of anticoagulation and 20-25% at 4 years, but there is an ~2% per year risk of major bleeding);3,5,19 the risks of death from a fatal pulmonary embolus or a major bleed are roughly equivalent. The patient was not considered to have the antiphospholipid antibody syndrome. Discussion ensued with the patient regarding enrollment in a long-term trial of low intensity anticoagulation (target INR 1.5-2) to prevent recurrent venous thromboembolism (the PREVENT trial); this is a double-blind trial in which half the enrolled patients receive placebo. Ultimately the patient opted against enrollment and the decision was made to resume chronic anticoagulation with a target INR of 2-3.
Case #6:
Symptomatic DVT following arthroscopic surgery: Is there any point in doing a “hypercoagulable workup”?
A 43-year-old male with no significant past medical history underwent left knee arthroscopic surgery in July 2001 for a meniscal tear. Following the procedure, he had the leg immobilized and 9 days later developed lower extremity pain and swelling. An ultrasound revealed evidence of a popliteal DVT. Laboratory evaluation on admission: normal CBC, PT, and aPTT. He was hospitalized for 2 days, received heparin, and was discharged on low molecular weight heparin and warfarin. He continued on warfarin with a target INR of 2-3 since the event and was seen in consultation as he had a number of questions regarding DVT and his management.
He has no previous history of DVT and denies any family history of blood clots or pulmonary emboli. He is otherwise in good health and denies any systemic complaints. He is an attorney, does not smoke, and drinks alcohol socially. Physical exam reveals 2+ pitting edema in the left lower extremity up to the knee. The right lower extremity is without any edema.
Discussion (Dr. Heit):
While the incidence of venographic DVT after arthroscopic knee surgery may be substantial, the incidence of symptomatic DVT is quite low.25 Therefore, I am suspicious that this patient has an underlying thrombophilia. Because the DVT was associated with a transient risk factor (e.g., surgery), the risk of unprovoked venous thromboembolism recurrence is low. Consequently, I would recommend a shorter duration of oral anticoagulation therapy (e.g., 3 months). I also would recommend that special coagulation testing be performed 1 month after the patient completes oral anticoagulation therapy because I believe case-finding of an inherited thrombophilia is important for counseling both the patient and the patient’s family.
An alternate viewpoint (Drs. Bauer and Rosendaal):
The major risk factor for DVT in this patient is the recent arthroscopic surgery and postoperative immobilization; 3 months of anticoagulation was recommended. While symptomatic proximal venous thrombosis is quite infrequent following arthroscopic surgery, venograms have documented asymptomatic thrombi, predominantly in the deep veins of the calf of the operated leg, in 8-17% of patients. As the presence of an underlying hereditary defect would not affect our management given the clear precipitant for DVT and the absence of a prior personal or familial history of thrombosis, no laboratory evaluation was undertaken. The possibility of developing postphlebitic syndrome was discussed with the patient and continued use of a compression stocking on the left lower extremity was recommended, especially during the day until the edema had resolved.
Risk factors for venous thrombosis.
| Acquired | Inherited | Mixed/Unknown |
|---|---|---|
| Abbreviations: TAFI, thrombin activatable fibrinolysis inhibitor; APC, activated protein C. | ||
| Age | Antithrombin deficiency | Hyperhomocysteinemia |
| Previous thrombosis | Protein C deficiency | High levels of factor VIII |
| Immobilization | Protein S deficiency | APC-resistance in the |
| Major surgery | Factor V Leiden | absence of factor V Leiden |
| Orthopedic surgery | Prothrombin 20210A | High levels of factor IX |
| Malignancy | Dysfibrinogenemia | High levels of factor XI |
| Oral contraceptives | High levels of TAFI | |
| Hormonal replacement therapy | ||
| Antiphospholipid syndrome | ||
| Essential thrombocythemia | ||
| Polycythemia vera | ||
| Paroxysmal nocturnal hemoglobinuria | ||
| Acquired | Inherited | Mixed/Unknown |
|---|---|---|
| Abbreviations: TAFI, thrombin activatable fibrinolysis inhibitor; APC, activated protein C. | ||
| Age | Antithrombin deficiency | Hyperhomocysteinemia |
| Previous thrombosis | Protein C deficiency | High levels of factor VIII |
| Immobilization | Protein S deficiency | APC-resistance in the |
| Major surgery | Factor V Leiden | absence of factor V Leiden |
| Orthopedic surgery | Prothrombin 20210A | High levels of factor IX |
| Malignancy | Dysfibrinogenemia | High levels of factor XI |
| Oral contraceptives | High levels of TAFI | |
| Hormonal replacement therapy | ||
| Antiphospholipid syndrome | ||
| Essential thrombocythemia | ||
| Polycythemia vera | ||
| Paroxysmal nocturnal hemoglobinuria | ||
Thrombosis risk in acquired risk situations: The Leiden Thrombophilia Study.*
| Risk Factor | Patients (n = 474) | Controls (n = 474) | OR | CI 95 |
|---|---|---|---|---|
| *Details of the Leiden Thrombophilia Study have been described in detail elsewhere.64 Briefly, it is a population based case-control study; cases were unselected consecutive patients aged 18-70 with a first objectively diagnosed deep-vein thrombosis and controls were acquaintances of cases or spouses of (other) cases. Controls were matched for age and sex. Subjects with active malignancies were excluded. | ||||
| Time window for surgery, hospitalization (without surgery), and immobilization (not in the hospital, >13 days) was 1 year preceding the index date (i.e., date of thrombosis diagnosis in patients, similar date in controls); for puerperium it was delivery 30 days or less prior to the index date; and for pregnancy and oral contraceptives it was at the index date. Data on pregnancy, puerperium, and oral contraceptive use refer to women of childbearing age; hence the denominator is not all 474 patients or controls. | ||||
| Abbreviations: OR, odds ratio; CI, confidence interval. | ||||
| n(%) | n(%) | |||
| Surgery | 85 (18) | 17 (3.6) | 5.9 | 3.4–10.1 |
| Hospitalization | 59 (12) | 6 (1.3) | 11.1 | 4.7–25.9 |
| Immobilization | 17 (3.6) | 2 (0.4) | 8.9 | 2.0–38.2 |
| Pregnancy | 8 (5.0) | 2 (1.3) | 4.2 | 0.9–19.9 |
| Puerperium | 13 (8.2) | 1 (0.6) | 14.1 | 1.8–109 |
| Oral contraceptives | 109 (70) | 65 (38) | 3.8 | 2.4–6.0 |
| Risk Factor | Patients (n = 474) | Controls (n = 474) | OR | CI 95 |
|---|---|---|---|---|
| *Details of the Leiden Thrombophilia Study have been described in detail elsewhere.64 Briefly, it is a population based case-control study; cases were unselected consecutive patients aged 18-70 with a first objectively diagnosed deep-vein thrombosis and controls were acquaintances of cases or spouses of (other) cases. Controls were matched for age and sex. Subjects with active malignancies were excluded. | ||||
| Time window for surgery, hospitalization (without surgery), and immobilization (not in the hospital, >13 days) was 1 year preceding the index date (i.e., date of thrombosis diagnosis in patients, similar date in controls); for puerperium it was delivery 30 days or less prior to the index date; and for pregnancy and oral contraceptives it was at the index date. Data on pregnancy, puerperium, and oral contraceptive use refer to women of childbearing age; hence the denominator is not all 474 patients or controls. | ||||
| Abbreviations: OR, odds ratio; CI, confidence interval. | ||||
| n(%) | n(%) | |||
| Surgery | 85 (18) | 17 (3.6) | 5.9 | 3.4–10.1 |
| Hospitalization | 59 (12) | 6 (1.3) | 11.1 | 4.7–25.9 |
| Immobilization | 17 (3.6) | 2 (0.4) | 8.9 | 2.0–38.2 |
| Pregnancy | 8 (5.0) | 2 (1.3) | 4.2 | 0.9–19.9 |
| Puerperium | 13 (8.2) | 1 (0.6) | 14.1 | 1.8–109 |
| Oral contraceptives | 109 (70) | 65 (38) | 3.8 | 2.4–6.0 |
Test ordering practices of the panel.
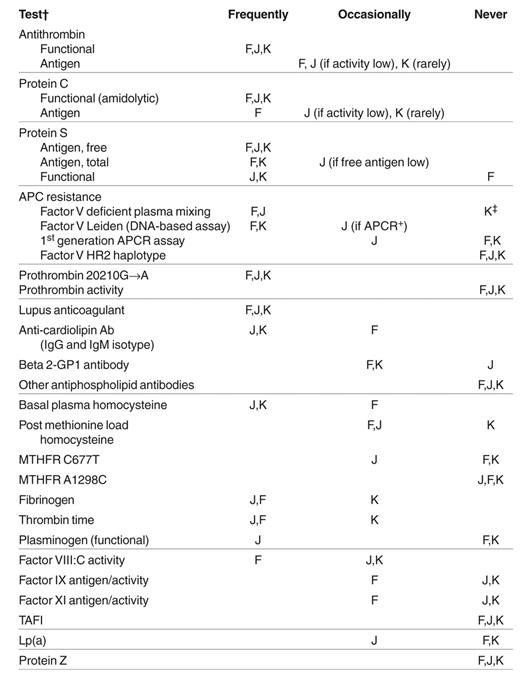
Abbreviations: F, Frits Rosendaal; J, John Heit; K, Ken Bauer; APC, activated protein C; APCR, APC resistance; MTHFR, methylene tetrahydrofolate reductase; TAFI, thrombin activatable fibrinolysis inhibitor.
†For F, no testing is done for a first episode of venous thrombosis unless it is exceptional in some way (e.g., very extensive clot, very young individual, positive family history). Thus, test-ordering practices refer to those exceptional first and recurrent events.
‡For K, hospital laboratories initially test for factor V Leiden by DNA-based assay.

Interaction of factor V Leiden and oral contraceptive use (left panel),89 and factor V Leiden and hormonal replacement therapy (right panel).90
The bars show the risk of those with only factor V Leiden, only oral contraceptives/hormonal replacement therapy, and those with both oral contraceptives/hormone replacement therapy and factor V Leiden, all relative to those with neither.
Interaction of factor V Leiden and oral contraceptive use (left panel),89 and factor V Leiden and hormonal replacement therapy (right panel).90
The bars show the risk of those with only factor V Leiden, only oral contraceptives/hormonal replacement therapy, and those with both oral contraceptives/hormone replacement therapy and factor V Leiden, all relative to those with neither.
