
Abstract
Over the last 2 decades, four major therapeutic approaches have drastically changed the prognosis in chronic myelogenous leukemia (CML): 1) allogeneic stem cell transplant (SCT); 2) interferon alpha (IFN-α) based regimens; 3) donor lymphocyte infusions (DLI); and 4) and the revolutionary BCR-ABL tyrosine kinase inhibitors such as STI571 (signal transduction inhibitor 571). Each modality has exploited and targeted different aspects of CML biology, and is associated with different risk-benefit ratios.
In Section I of this review, Dr. Melo reviews the molecular pathophysiology of CML and potential new targets for therapy including anti-sense strategies to disrupt the BCR-ABL gene and inhibition of the BCR-ABL tyrosine kinase activity.
In Section II, Dr. Tura, addresses important questions in the use of IFN-α for the treatment of CML, including the mechanism of action and the development of resistance, the optimal dose and duration of therapy and the prediction of response based on clinical features. An approach to the choice of therapy based on the predicted mortality is presented.
In Section III Dr. Giralt presents an update on the results of unrelated donor transplantion, donor lymphocyte infusions (DLI) and non-ablative stem cell transplantation (NST) in CML. The roles of CD8-depletion, dose escalation and the transduction of suicide genes in treatment with DLI are addressed. Early results of NST in CML show that it is feasible and can result in long-term disease control.
In Section IV Drs. Kantarjian and Talpaz review the results of IFN-α plus low-dose cytosine arabinoside and other promising modalities for CML including homoharringtonine, decitabine, and polyethylene glycol-interferon. In Section V they present an update on the recent experience with STI571. Objective but transient responses have been seen in 40% to 50% of patients in CML blastic phase. In accelerated phase, the response rate with STI571 exceeds 70%, and these responses are durable. In chronic phase CML, STI571 at 300 mg daily in patients who failed IFN-α produces a complete hematologic response (CHR) in over 90% of patients. Early results suggest cytogenetic response rates of approximately 50%, which may be major in approximately 30%. The maturing results with STI571 may soon change current recommendations regarding the relative roles of established modalities such as allogeneic SCT and IFN-α. Important questions include 1) whether STI571 therapy alone may be sufficient to induce long-term survival and event-free survival in CML, or whether it needs to be combined simultaneously or sequentially with IFN-α and cytosine arabinoside; and 2) what should the indications for frontline allogeneic SCT be in relation to STI571 therapy.
I. CML: Molecular Pathophysiology and Potential New Targets for Therapy
Junia V. Melo, M.D., Ph.D.*
Department of Hematology—RPMS, Hammersmith Hospital, Ducane Road, London W12 0NN, UK
CML was the first neoplastic process to be linked to a consistent acquired genetic abnormality, and it is by now the best studied molecular model of leukemia. This knowledge, together with the availability of sophisticated biochemical and biophysical technology, offers a unique opportunity to develop rational molecularly targeted therapies for this leukemia.
The Molecular Pathophysiology of CML
The crucial genetic event in CML is the generation of a t(9;22)(q34;q11) reciprocal chromosomal translocation in a hematopoietic stem cell. This translocation creates two new genes, BCR-ABL on the 22q- or Ph chromosome, and the reciprocal ABL-BCR on the derivative 9q+. The latter gene, although transcriptionally active,1 does not appear to have any functional role in the disease.
Depending on the breakpoint in the BCR gene, three main types of BCR-ABL gene can be formed.2 The predominant hybrid gene in classical CML is derived from a disruption in the major breakpoint cluster region (M-bcr). Transcription from this gene yields chimeric mRNA molecules where the fusion between BCR and ABL sequences is represented by a b3a2 and/or a b2a2 junction (Figure 1). The final product of this genetic rearrangement is a 210 kDa cytoplasmic fusion protein, or p210BCR-ABL, the oncoprotein responsible for most, if not all, phenotypic abnormalities of chronic phase CML. Much more rarely, CML can result from hybrid genes derived from breaks in the minor (m)-bcr (with e1a2 transcripts) or in the micro (m)-bcr (e19a2 transcripts) regions. The disease in these cases may have particular phenotypic features, such as significant monocytic or neutrophilic/thrombocythemic (p230CML) components.3,4,5

Structure of the genomic BCR and ABL loci with their respective breakpoint cluster regions (M-bcr in BCR and those indicated by vertical arrows in ABL), of the chimeric BCR-ABL mRNA transcripts (with b3a2 or b2a2 junctions), and of the p210BCR-ABLfusion protein characteristic of chronic myelogenous leukemia (CML).
Some of the important functional domains of this protein are illustrated, such as the oligomerization domain, the tyrosine 177 (Grb-2 binding site), the phospho-serine/threonine (P-S/T)-rich SH2-binding domain and the rho-GEF (dbl-like) domain on the Bcr portion; and the regulatory src-homology regions SH3 and SH2, the SH1 (kinase domain) with its main site of autophosphorylation (Y412), the nuclear localization signal (NLS), and the DNA- and actin-binding domains in the Abl moiety. Several of these domains may be targeted therapeutically with the objective of inhibiting the transforming activity of the Bcr-Abl oncoprotein.
Structure of the genomic BCR and ABL loci with their respective breakpoint cluster regions (M-bcr in BCR and those indicated by vertical arrows in ABL), of the chimeric BCR-ABL mRNA transcripts (with b3a2 or b2a2 junctions), and of the p210BCR-ABLfusion protein characteristic of chronic myelogenous leukemia (CML).
Some of the important functional domains of this protein are illustrated, such as the oligomerization domain, the tyrosine 177 (Grb-2 binding site), the phospho-serine/threonine (P-S/T)-rich SH2-binding domain and the rho-GEF (dbl-like) domain on the Bcr portion; and the regulatory src-homology regions SH3 and SH2, the SH1 (kinase domain) with its main site of autophosphorylation (Y412), the nuclear localization signal (NLS), and the DNA- and actin-binding domains in the Abl moiety. Several of these domains may be targeted therapeutically with the objective of inhibiting the transforming activity of the Bcr-Abl oncoprotein.
The Bcr-Abl protein is leukemogenic because its Abl-derived tyrosine kinase is constitutively activated. Unlike the situation of the normal Abl protein, whose tyrosine kinase activity is tightly regulated, Bcr-Abl transduces signals to various pathways in an autonomous fashion. This leads to malignant transformation by interference with basic cellular processes, such as control of proliferation,6 adherence,7 and physiological death.8 Mutational analyses have identified several features or domains in the Bcr-Abl fusion protein that are essential for cellular transformation. In the Abl moiety they include the SH1 (tyrosine kinase), SH2 and actin-binding domains, and in the Bcr portion a coiled-coil oligomerization domain contained in amino acids (aa) 1-63, the tyrosine at position 177 and phosphoserine/threonine rich sequences between aa 192-242 and 298-413.
A large number of substrates can be phosphorylated by Bcr-Abl (Figure 2). Due to autophosphorylation there is a marked increase of phosphotyrosine on Bcr-Abl itself, which creates binding sites for the SH2 domains of other proteins. Generally, substrates of Bcr-Abl can be grouped according to their physiological role into adaptor molecules (such as Grb-2, Crkl and Dok), proteins associated with the organization of the cytoskeleton and the cell membrane (such as paxillin, talin and Fak) and a third group of proteins with catalytic function (such as the non-receptor tyrosine kinase Fes, PI-3 kinase and the phosphatase Syp). The choice of substrates depends on the cellular context. For example, Crkl is the major tyrosine phosphorylated protein in CML neutrophils, whereas phosphorylated Dok is predominantly found in early progenitor cells.

Pathways involved in Bcr-Abl signalling.
A large number of Bcr-Abl substrates and associated proteins have been identified, a selection of which are shown here. For some of these both the active (solid) and the inactive (punctuated) forms are illustrated. Inhibition of individual pathways by targeting proteins downstream of Bcr-Abl (e.g. Grb-2, Ras, PI-3 kinase) may be effective to suppress individual aspects of the CML cell phenotype, although the redundancy of the system may work against the therapeutic success of such strategies.
Pathways involved in Bcr-Abl signalling.
A large number of Bcr-Abl substrates and associated proteins have been identified, a selection of which are shown here. For some of these both the active (solid) and the inactive (punctuated) forms are illustrated. Inhibition of individual pathways by targeting proteins downstream of Bcr-Abl (e.g. Grb-2, Ras, PI-3 kinase) may be effective to suppress individual aspects of the CML cell phenotype, although the redundancy of the system may work against the therapeutic success of such strategies.
The signalling network controlled by Bcr-Abl is complex and highly redundant (Figure 2). The sum of these protein interactions translates into the altered phenotype of CML cells, which consists of constitutively active mitogenic signalling, defective adherence to stromal cells and extracellular matrix, and reduced response to apoptosis-inducing stimuli. `Correction' of any of these defects by a rationally designed therapeutic tool would very likely tip the balance towards the re-establishment of normal hematopoiesis.
Possible Levels of Intervention
Prevention of CML
Prevention of CML would be better than cure. Unfortunately, except for its association with previous exposure to high-dose ionizing radiation, no significant risk factor has been associated with the development of CML. Recent studies on the mechanisms of leukemia-associated chromosomal translocations have shown that BCR-ABL, as well as other transcriptionally functional genes, is generated spontaneously in hematopoietic cell lines in culture9 and, even more importantly, in circulating normal leukocytes from healthy adults.10,11 This low level of background translocations is part of the intrinsic instability of the human genome, and is not currently preventable.
In theory, one could prevent the development of CML by attempting to increase the immunological surveillance against BCR-ABL-containing cells through vaccination with Bcr-Abl peptides (see below). Because of the low frequency of CML, the cost-effectiveness of such undertaking would be unacceptable, unless a specific group of individuals at higher risk of developing CML could be identified.
Disruption of BCR-ABL
Once a `successful' t(9;22) is perpetuated in an expanding clone of hematopoietic cells, chronic phase CML develops. At this stage, the only known identified genetic abnormality is the BCR-ABL gene itself, although other as yet unidentified mutation(s) may have preceded and facilitated the emergence of the BCR-ABL-containing clone. The BCR-ABL gene and its cognate mRNA and fusion protein are in principle ideal targets for disruption in an attempt to remove the neoplastic machinery of the leukemia cells. Strategies aimed at blocking BCR-ABL at different levels of expression are at the forefront of current investigations.
Disruption of the Bcr-Abl-associated pathway
An alternative to neutralizing Bcr-Abl would be to target proteins that are directly or indirectly modulated by Bcr-Abl in its various oncogenic pathways. Since the effects of Bcr-Abl are multifactorial, inhibition of one pathway may not be sufficient to abrogate effectively the malignant phenotype.
Molecularly Designed Strategies
Molecularly designed strategies, which are based on our current knowledge of the molecular and cell biology of CML, have concentrated on three main areas: (a) inhibition of gene expression at the translational level by `antisense' strategies; (b) modulation of protein function by specific signal transduction inhibitors; and (c) stimulation of the immune system's capacity to recognize and destroy the leukemic cells.
Antisense strategies
Antisense RNA and DNA oligonucleotides (ODNs) targeted against nuclear genomic DNA or cytoplasmic mRNA, synthetic DNA transcription factor `decoys,' ribozymes (catalytic RNA molecules)12 and DNA-binding proteins have all been employed as tools for targeting genetic defects in CML cells.13 In the traditional approach the exogenous nucleotide sequence hybridizes to the target mRNA creating a duplex that in effect forms a `jam' that prevents the ribosomal complex from reading along the message. The antisense strategies received much attention in the last decade but, due to a number of technical problems, have in general failed to fulfill their theoretical promises (recently reviewed in reference14 ). New modifications to the system, however, such as the use of BCR-ABL junction-specific catalytic subunits of RNase P may revitalise the field.15
Although BCR-ABL may in theory be the most attractive target for antisense therapy, the long half life (in excess of 24 hours) of its protein poses a significant obstacle. This led some investigators to investigate antisense ODNs against downstream targets of Bcr-Abl, such as MYC, CRKL, GRB2, KIT, VAV and MYB. Of these, only anti-MYB ODNs have been tested in clinical trials, but long-term follow-up results of these trials have not been reported.16
Signal transduction inhibitors
If translation of Bcr-Abl and related proteins cannot be adequately halted by an antisense technology, the next level for molecular intervention is to block protein function. Preventing a protein from exerting its role in the oncogenic pathway should ultimately lead to elimination of the leukemic phenotype.
The development of signal transduction inhibitors (STIs) has evolved in close parallel to the unravelling of the biochemical properties of proteins involved in the signalling network in hematopoietic cells. These inhibitors usually target a specific functional domain of a protein. As with the antisense technology, the ideal STI for the treatment of CML should be able to block the Bcr-Abl fusion protein itself, as this would limit its `damaging' effects (e.g. anti-proliferative, pro-apoptotic, enhancement of cell adherence) to the cell clone that needs to be `damaged.' In practice, though, inhibition of selected Bcr-Abl-associated proteins may be just as effective in disrupting the oncogenic process in the leukemia cell, with little or no adverse consequence to the normal tissues where the given protein is expressed under physiological control.
a) Inhibitors of the Bcr-Abl fusion protein: Since the main transforming property of Bcr-Abl is exerted through its constitutive tyrosine kinase activity, direct inhibition of such activity seems to be the most logical means of `silencing' the oncoprotein. To this effect, several tyrosine kinase inhibitors have been evaluated for their potential to modify the phenotype of CML cells. The first to be tested were compounds isolated from natural sources, such as the isoflavonoid genistein and the antibiotic herbimycin A.17 Later, synthetic compounds known as tyrphostins were developed through a rational design of chemical structures capable of competing with the ATP or the substrate for the binding site in the catalytic center of the kinase.18
One of these tyrphostins (AG1112), genistein, and herbimycin A were variably reported to inhibit the p210BCR-ABL tyrosine kinase, to induce differentiation of the K562 CML cell line and to inhibit the in vitro growth of some BCR-ABL-positive cell lines. Genistein has also been shown to exert a strong anti-proliferative effect on CML colony-forming progenitor cells, whereas AG957 was observed to restore β1-integrin-mediated adhesion and inhibitory signalling in CML progenitors.
So far, the most successful of the molecularly designed ATP competitors is the 2-phenylaminopyrimidine STI571 (previously known as CGP57418B), from Novartis Pharma (Basel, Switzerland), which specifically inhibits Abl tyrosine kinase at submicromolar concentrations.19 Inhibition of the Bcr-Abl kinase activity by this compound results in transcriptional modulation of various genes involved in the control of the cell cycle, cell adhesion and cytoskeletal organization, leading the Ph-positive cell to an apoptotic death.20 STI571 selectively suppresses the growth of CML primary cells and cell lines both in vitro and in mice.21,22,23 As described later in this review, this compound is now being tested in several clinical trials, with exceptionally good results.
To anticipate and possibly overcome CML resistance to STI571, investigators have generated murine and/or human BCR-ABL-positive cell lines resistant to STI571. These have shown that the most frequent mechanism of resistance is amplification and overexpression of the BCR-ABL gene.24 Overexpression of the Pgp glycoprotein, the product of the multidrug resistance (MDR) gene, may also contribute to the resistant phenotype. However, STI571-resistant clones from some cell lines, e.g. KCL22, do not show either of these resistance phenotypes, suggesting that resistance to STI571 may evolve by multiple mechanisms. Patients in lymphoid blast crisis have a rapid and significant response to treatment with STI571 but relapse after a median time of 10.5 weeks. It will be important to study the causes of resistance in these primary CML cells, in order to design modifications to the treatment. It is possible that combinations of STI571 with other drugs such as interferon-α (IFN-α), daunorubicin or cytosine arabinoside (ara-C) may be more effective than STI571 alone, as suggested by in vitro studies of cell lines and primary CML cells.25
Two possible molecular side effects of Abl tyrosine kinase inhibitors should be kept in mind. The first are the consequences of inhibition of the normal Abl kinase activity, which may be related to the regulation of cell proliferation and lymphoid development and, if suppressed, may diminish cellular immunity. Bcr-Abl/Abl tyrosine kinase inhibitors may also inhibit other tyrosine kinases, namely the PDGF receptor and the steel factor receptor (c-kit), potentially interfering with normal cellular functions. The negligible degree of side effects observed in the STI571 clinical trials so far suggests that if suppression of the normal Abl and other tyrosine kinases occurs, it may be compensated by activation of alternative pathways.
In addition to the tyrosine kinase, inhibition of other relevant domains of p210BCR-ABL may have a negative effect on the survival and proliferative potential of Ph-positive cells. Candidates for manipulation are the Bcr oligomerization domain and the Abl SH2 and actin-binding domains. Inhibitors being tested are short peptides designed to be homologous to all or part of a given domain, which compete with p210BCR-ABL for ligands or substrates important for the transforming capacity of the full-length protein. These inhibitors are administered to the cell either via transfection of a recombinant expression vector encoding the designed peptide, or as a synthetic drug acting as a peptidomimetic.26 Arlinghaus and co-workers27 have shown that adenovirus-mediated transfer of a mutant Bcr 64-413 (containing the first exon minus the oligomerization domain) into the K562 and BV173 CML cell lines inhibited Bcr-Abl kinase activity and colony formation. Introduction of this Bcr fragment into bone marrow cells from a CML patient suppressed their proliferation in suspension culture and promoted their adhesion to the culture flask, suggesting that this treatment may correct some aspects of the CML phenotype.
b) Inhibitors of other signal transduction proteins: The use of farnesyl transferase inhibitors (FTIs) against BCR-ABL-induced leukemias is starting to gain momentum. The rationale for this strategy was again based on the notion that Bcr-Abl signals via activation of the Ras/MAPK pathway. In order to be activated, the Ras protein has to be anchored to the cytoplasmic face of the plasma membrane through a covalently attached prenyl (usually a farnesyl) group. FTIs inhibit the 3-hydroxymethyl-glutaryl-coenzyme A reductase, an early and rate-limiting enzyme in the sterol synthesis pathway, thus depleting the cell of farnesyl pyrophosphate. Although it was initially thought that the anti-proliferative capacity of some FTIs was due to reduced post-translational farnesylation of Ras and consequent prevention of its membrane localization, it appears that other mechanisms, such as the gain of geranylgeranylated Rho-B, are also involved. It should be emphasized that current FTIs may not inhibit farnesylation of ras, or ras itself, although they may inhibit CML proliferation through other potential mechanisms. Encouraging preliminary results have been reported on inhibition of in vitro proliferation of ALL and juvenile myelomonocytic leukemia cells by these compounds.28 Investigations on the effect of FTIs in BCR-ABL-positive cells are warranted.
Another promising approach is the use of PI-3 kinase inhibitors. PI-3 kinase functions downstream of Bcr-Abl may involve both apoptosis and control of cytoskeleton organization. Treatment of BCR-ABL transfected cell lines with wortmannin, a potent PI-3 kinase inhibitor, normalizes their pattern of stress fibers and focal adhesion contacts and decreases their excessive spontaneous motility. When tested on CML cell lines and primary bone marrow CD34+ cells, wortmannin led to suppression of colony formation, with apparently no effect on colony formation by normal CD34+ cells.29 The next step is to test the efficacy and in vivo toxicity of the PI-3 kinase inhibitors in animal models of CML.
Immunological stimulation
The capacity of donor lymphocyte infusions (DLI) to induce complete remission, without cytotoxic chemotherapy, in a high proportion of patients who relapse after SCT is probably the best evidence for the role of the immunological pathway in CML. The mechanism is likely to be, at least in part, related to cytotoxic T cell lymphocytes (CTLs) that recognise CML targets. Major efforts have been devoted for the past 5 years to establishing an efficient strategy to enhance CML-specific T cell-mediated immunity.
In the post-allogeneic stem cell transplant setting, CTLs of donor origin can be generated in vitro when mixed with the recipient's leukemic cells.30 The stimulant in this case is either a leukemia-associated antigen (e.g. a product of the p210BCR-ABL or a proteinase 3 peptide)31 or minor histocompatibility antigens.32 Whatever the antigenic specificity, CTLs recognize targets in the context of major histocompatibility complex (MHC). The main obstacles in the development of CTL clinical protocols are the difficulties in generating specific lines or clones that can be expanded in vitro to the large quantities needed for effective therapy. Furthermore, the requirement for CTLs to be self-MHC restricted implies that not all patients would be eligible for this form of treatment.
An ingenious strategy has been recently tested for raising CTL against the Wilms' tumor (WT1) antigen, which is reported to be preferentially expressed in CML, as compared to normal CD34+ cells.33 In this case, WT1 peptide-specific CTL clones isolated from HLA-A0201- or HLA-A24-negative donors were able to kill leukemic cell progenitors from the respective HLA-A-positive counterpart patients, suggesting that selection of such lymphocytes for CTL adoptive immunotherapy may be used in a one-locus HLA-mismatch transplant protocol.
It is not yet clear how easily CTLs can be raised in the autologous setting. The fact that a clone of BCR-ABL-positive cells was allowed to expand and produce the disease in the first place suggests that some form of tolerance to the abnormal cells or immunosuppression must occur in these patients.
A new approach to induce a CTL response is to pulse accessory cells with exogenous 8-25 aa-long peptides derived from the b3a2 or b2a2 junctional regions of p210BCR-ABL. This approach was pioneered by Scheinberg et al, who conducted a phase I clinical trial in which the safety and immunogenicity of a multidose, multivalent b3a2 peptide vaccine were evaluated in 12 patients with CML in chronic phase. No significant adverse effects were seen. Three out of six patients treated at the two highest dose levels of vaccine generated peptide-specific T cell proliferative responses ex vivo and/or delayed type hypersensitivity responses, lasting up to 5 months after vaccination, although CTLs were not identified.34 The overall results suggest that a Bcr-Abl-derived peptide vaccine can be safely administered to CML patients and can elicit a specific immune response. It remains to be seen whether this type of vaccination will result in significant clinical benefit. Combining such molecularly oriented therapies (e.g. STI571 and FTIs) may synergize anti-CML efficacy and allow successful strategies in CML outside the setting of allogeneic SCT.
Summary and Conclusions
The considerable expansion of our knowledge of the molecular pathophysiology of CML has provided important new insights into targeting the treatment of patients to specific molecular defects. The ideal candidate for disruption should be the BCR-ABL gene and encoded fusion protein, since these are the molecules that are uniquely present in the CML cells. Thus, various strategies aim at neutralizing BCR-ABL proper, such as antisense ODNs or ribozymes against the junctional sequences, inhibition of Bcr-Abl tyrosine kinase activity by STIs, or enhancement of an immune response to BCR-ABL-containing cells. Alternative approaches are designed to block molecules that are constitutively activated by Bcr-Abl (e.g. Ras, PI-3 kinase, Grb2, Crkl). The ongoing search for proteins and pathways associated with the abnormal phenotype of the CML cell will surely uncover new molecular targets for a rational therapy of CML in the near future.
II. Important Questions Related to Interferon-α Therapy in Chronic Myeloid Leukemia
Sante Tura, M.D.*
University of Bologna, Inst. of Hematology and Oncology, Via G. Massarenti, 9, 40138 Bologna, Italy
IFN-α has become the treatment of choice in patients with Ph-positive CML who are not candidates for allogeneic stem cell transplant (SCT). It induces durable cytogenetic responses and prolongs duration of chronic phase and survival compared with conventional chemotherapy.35,36,37,38,39,40,41 Questions regarding IFN-α include a) the mechanisms of action of IFN-α; b) the best IFN-α dose schedule and duration of treatment; c) the optimal IFN-α combinations; d) the long-term outcome of patients on IFN-α therapy; e) the relationships between features of CML and probability of response to IFN-α and expected benefits; and f) definitions of clinical and biological targets of treatment, including the concept of cure.
The molecular and biologic bases of the mechanisms of action of IFN-α are poorly understood.35,42 The effect of IFN-α may be a direct anti-proliferative effect or an indirect one on the immune system through non-specific enhancement of anti-leukemic cell-mediated response. IFN-α may increase HLA molecule expression on Ph-positive cells, so that the HLA-linked leukemic peptide can be recognized more efficiently by antigen presenting cells and by T-lymphocytes.43,44 Binding of IFN-α to its membrane receptor activates a number of signalling pathways that eventually modulate the transcription of several genes.42 Many of these pathways are shared with those that are constitutively activated by the leukemia-specific BCR-ABL tyrosine-kinase oncoprotein p210. One important difference is ICSBP, which may be down-regulated in BCR-ABL-expressing cells and up-regulated by IFN-α. Mice lacking ICSBP expression develop a myeloproliferative syndrome resembling chronic phase CML. Why some Ph-positive cells are or become resistant to IFN-α is unknown. An indirect answer to the question is provided by the clinical observation that the therapeutic effect of IFN-α decreases with time, since it is much greater in early than in late chronic phase and is minimal with progression from chronic to accelerated or blastic phases.35,36 This implies the development of other genomic abnormalities that make the cells more resistant to IFN-α and perhaps to other modalities, including allogeneic SCT, homoharringtonine (HHT) and STI571. Sensitivity to IFN-α may be primarily dependent on p210 amount.45 The identification of other genomic abnormalities may provide a basis for a more rational use of IFN-α, either alone or in combination.
IFN-α was initially administered at a dose of about 5 million units (MU)/m2/daily, although most studies ultimately deliver a dose of 5 MU daily or less. The optimal IFN-α dose, particularly in combinations, is under investigation. Arguments regarding IFN-α dose are not always based on biologic and pharmacokinetic data, but on the fact that IFN-α is expensive, that its side effects are dose related (especially in the elderly), and that some patients respond well to lower doses of IFN-α. Two prospective studies in the UK and in the Netherlands are now comparing low-dose (3 MU five times a week) versus high-dose (5 MU/m2 daily). These studies address treatment toxicity and cost-effectiveness, not improved efficacy, which is the major drawback of IFN-α. The activity of low-dose IFN-α may provide important information for the combination of IFN-α with other agents.
A third important question is whether IFN-α therapy can cure CML. Cure in CML can be considered clinical, (i.e. resulting in durable hematologic and cytogenetic remissions) or biological (resulting in molecular evidence absent of very limited residual disease).46,47,48 Current molecular techniques can identify as few as 30 or 40 molecules of bcr-abl transcript and detect one Ph-positive cell in a million cells. The concept of cure in CML and the role of molecular biology in providing guidelines for treatment have been discussed extensively.49,50 Data on patients who achieve a complete cytogenetic (CG) response (CG CR) may provide valuable information. Since the frequency of CG CR ranges between less than 10% and 35%, European investigators have gathered cases of Ph-positive CML who had achieved a CG CR at least once on IFN-α alone. This registry of CG CR includes now 317 cases, seen between 1983 and 1997, for an observation time ranging between 2 and 15 years (median 6.5 years). Their median age is 49 years (range 9 to 73 years). The percentage of low-risk patients is 62% using Sokal's risk criteria51 and 58% using the new Euro risk criteria.52 At last follow-up, 212/316 patients (67%) were still in CG CR, 36 (11%) were in major or partial cytogenetic response, 30 (9%) had lost their cytogenetic response but were still in hematologic remission, 5 (2%) had died in chronic phase, 5 (2%) had undergone transplantation, and only 28 (9%) had progressed to accelerated or blastic phase. The 10-year projected survival was more than 80% from diagnosis and more than 70% from date of fist CG CR. The projected 10-year survival was more than 90% for low-risk patients, 75% for intermediate-risk patients, and only 50% for high-risk patients. This suggests that long-term survival (potential cure) is possible in low-risk patients.
Two recent analyses compared cohorts of patients treated with IFN-α versus allogeneic SCT.54,57 In the Italian experience comparing IFN-α versus allogeneic SCT in CML, the authors concluded that initial allogeneic SCT may be superior among younger patients (age ≤ 32 years) and those with higher risk disease, while initial IFN-α therapy may be superior among older patients and those with low-risk disease.54 When patients were cross tabulated into four categories by age (32 years or younger versus older) and risk group (higher versus low), only patients who were 32 years old or younger and who had higher-risk disease (61/422 = 14% of total) had a significant 10-year survival benefit with allogeneic SCT versus IFN-α (10-year survival rate 65% versus 14%, p =.008). Patients older than 32 years with higher risk disease showed a similar trend (10-year survival rate 47% versus 17%; p =.50), which was perhaps not significant because only 32 patients had undergone allogeneic SCT while 130 had received IFN-α. However, in low-risk patients (219/422 = 52% of total), there was no survival advantage for one modality over another. Hehlmann et al compared the outcome of allogeneic SCT versus IFN-α based on “genetic randomization,” i.e. patients were allocated according to eligibility for transplant and then received allogeneic SCT if available. Of 901 patients, 524 were genetically randomized: 165 (18% of total) for SCT and 389 for IFN-α therapy. Median observation time was 639 days. During the first 4 years of observation survival was better with IFN-α (p = 0.014), but the survival curves were expected to cross at 5 years. Early mortality from SCT was 25% from unrelated donors and 30% from related donors. While the authors suggest that SCT outcome will be more favorable with longer follow-up among intermediate-high risk patients, this is not observed for low-risk patients, although they speculate it may occur with much longer follow-up.57
The long-term observations of patients who achieved CG CR with IFN-α alone and of the outcome of IFN-α versus allogeneic SCT provide some guidelines for the treatment of CML with IFN-α based on response and risk.53,54 Low-risk patients constitute about 50% of those less than 55 years old and about 30% of older patients. A guideline is shown in Table 1. Patients younger than 30 years should be considered for frontline allogeneic SCT, regardless of risk group. Among older patients, low risk patients are likely to respond to IFN-α and those who achieve a CG CR can survive for prolonged periods of time. They are the best candidate for IFN-α treatment. In contrast, non-low risk patients may respond to IFN-α and survive longer, but their survival is not prolonged indefinitely. For the patients who are less than 55 years old, the controversial question of IFN-α therapy versus allogeneic SCT upfront depends on the probability of cure and the risk of transplant-associated mortality53,54,55 (Table 2).
The guidelines for the treatment of chronic myelogenous leukemia (CML) with interferon-α (IFN-α) or allogeneic stem cell transplantation (SCT) are based on risk, age and the availability of a suitable marrow donor.
| Testing for IFN sensitivity may help decide between IFN-α and allogeneic SCT. Testing for IFN-α sensitivity may require some time, but treatment of low-risk patients is never urgent. In the table, the term investigational treatment encompasses a number of procedures (e.g. intensified chemotherapy with autologous stem cell rescue, allogeneic SCT from mismatched donors or in the elderly) and novel agents. However, the first-line investigational option to be considered should be the BCR-ABL tyrosine-kinase inhibitor STI571, either alone or in combination. |
| Age < 30 years, any risk |
|
| Age 30-55 years, low risk |
|
| Age 30-55 years, non-low risk |
|
| Age > 55 years, low risk |
|
| Age > 55, non-low risk |
|
| Testing for IFN sensitivity may help decide between IFN-α and allogeneic SCT. Testing for IFN-α sensitivity may require some time, but treatment of low-risk patients is never urgent. In the table, the term investigational treatment encompasses a number of procedures (e.g. intensified chemotherapy with autologous stem cell rescue, allogeneic SCT from mismatched donors or in the elderly) and novel agents. However, the first-line investigational option to be considered should be the BCR-ABL tyrosine-kinase inhibitor STI571, either alone or in combination. |
| Age < 30 years, any risk |
|
| Age 30-55 years, low risk |
|
| Age 30-55 years, non-low risk |
|
| Age > 55 years, low risk |
|
| Age > 55, non-low risk |
|
Proposed risk classification for allogeneic stem cell transplantation (SCT).55
| Risk factors are donor type (0 for HLA-identical sibling donor, 1 for a matched unrelated donor); disease stage (0 for first chronic phase, 1 for accelerated phase, and 2 for blast crisis or higher chronic phase); age of recipient (0 for < 20 years, 1 for 20-40 years, and 2 for > 40 years); sex combination (0 for all, except 1 for male recipient/female donor); and time from diagnosis to SCT (0 for < 12 months, 1 for > 12 months) | |||
| Results at 5 years | |||
| Risk Factors | % of Patients with Risk Factors | Leukemia-Free Survival (%) | Transplant-Related Mortality (%) |
| 0 | 2 | 60 | 20 |
| 1 | 18 | 60 | 23 |
| 2 | 28 | 47 | 31 |
| 3 | 28 | 37 | 46 |
| 4 | 15 | 35 | 51 |
| 5 | 7 | 19 | 71 |
| 6-7 | 2 | 16 | 73 |
| Adapted from Gratwohl et al.55 | |||
| Risk factors are donor type (0 for HLA-identical sibling donor, 1 for a matched unrelated donor); disease stage (0 for first chronic phase, 1 for accelerated phase, and 2 for blast crisis or higher chronic phase); age of recipient (0 for < 20 years, 1 for 20-40 years, and 2 for > 40 years); sex combination (0 for all, except 1 for male recipient/female donor); and time from diagnosis to SCT (0 for < 12 months, 1 for > 12 months) | |||
| Results at 5 years | |||
| Risk Factors | % of Patients with Risk Factors | Leukemia-Free Survival (%) | Transplant-Related Mortality (%) |
| 0 | 2 | 60 | 20 |
| 1 | 18 | 60 | 23 |
| 2 | 28 | 47 | 31 |
| 3 | 28 | 37 | 46 |
| 4 | 15 | 35 | 51 |
| 5 | 7 | 19 | 71 |
| 6-7 | 2 | 16 | 73 |
| Adapted from Gratwohl et al.55 | |||
These guidelines are based on the results of IFN-α therapy alone and on the current accepted risks of transplant in different patient subsets (based on age, related versus unrelated donors, degree of mismatching, and patient selection). These guidelines, including the roles of frontline allogeneic SCT and of IFN-α therapy, should be re-evaluated as data with STI571 matures.21,22,23 ,56 Future investigations of STI571 alone or combined with IFN-α, ara-C, HHT, decitabine, or other molecular strategies (e.g. FTIs) may provide superior survival and durable complete and major cytogenetic responses. Guidelines would then be based, not on the comparative results of allogeneic SCT versus IFN-α, but on the SCT-associated mortality (i.e. the acceptable upfront risk for an established curative modality). A reasonable approach would be as follows:
If SCT-associated one-year mortality is below 20% (younger patients with matched-related donors), allogeneic SCT is the frontline treatment option.
If mortality is between 20% to 40% (older patients up to 50 years of age with related donors, or young patients with matched unrelated donors), only patients who fail therapy (lack of cytogenetic response after one year of therapy, subsequent loss of cytogenetic response) are offered allogeneic SCT in chronic phase.
If mortality is estimated to exceed 40%, investigational options are offered in chronic phase (HHT, decitabine, tyrosine kinase inhibitors) and allogeneic SCT proposed if signs of disease transformation appear. Patients may then survive many years in chronic phase. Furthermore, the transplant-related mortality in accelerated phase is not sufficiently higher in these patients to justify the upfront high risk of transplant mortality in chronic phase.
III. Updates On Unrelated Donor Transplantation, Donor Lymphocyte Infusions, And Non-Ablative Stem Cell Transplantation
Sergio Giralt, M.D.*
M.D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box 65, Houston TX 77030-4009
Unrelated Donor Transplantation
Only 35% of patients with CML under the age of 55 actually undergo an allogeneic transplant, primarily due to the lack of an appropriate tissue-compatible related donor.58 Alternative progenitor cell sources such as cord blood, mismatched related transplants or matched unrelated donor transplants have been extensively explored as treatment options for patients with CML. CML is the most common indication for unrelated donor transplants and represents 35% of all unrelated donor transplants done in the United States.59 With the increasing number and size of volunteer donor registries, the median time for identification of a suitable donor has decreased from 6.9 months to 5.5 months; however, only one of four searches actually culminates in transplantation.60 Results of the largest series of unrelated donor transplantation for CML are summarized in Table 3.59 ,61,62,63
Results of representative studies of unrelated donor transplantation for chronic myelogenous leukemia (CML).
Acute graft-versus-host disease (GVHD) and infectious complications remain the most important causes of morbidity and mortality of unrelated donor transplantation.59,60,61,62,63,64 The single most important prognostic factor for development of acute GVHD is degree of histocompatibility. Serologic typing underestimates the degree of mismatching in a large proportion of donor-recipient pairs. Modern molecular techniques have demonstrated that molecular mismatches in serologically matched donor-recipient pairs are common and correlate with transplant outcomes.65,66,67 In the Seattle study, young patients with CML (less than 50 years of age), who receive a fully molecular matched unrelated donor and are transplanted within 1 year of diagnosis have an estimated 5-year survival rate of 74%, indistinguishable from recipients of fully matched sibling donor transplants.62 The impact of relevant pre-treatment variables on outcome of unrelated donor transplants in CML is summarized in Table 4.
Variables that affect outcomes of unrelated donor transplants in chronic myelogenous leukemia (CML).
| Outcome | ||
|---|---|---|
| Variable (Reference) | GVHD | Survival |
| Chronic phase status at time of SCT (59,62) | - | + |
| Serologic mismatch Class I or II (59,61,65) | + | 0 |
| Molecular mismatch (62,66,67) | + | - |
| Older age (59,62) | 0 | - |
| Transplant < 12 months from diagnosis (59,62) | 0 | + |
| CMV positivity (59,61) | 0 | - |
| T-cell depletion (59) | - | 0 |
| Abbreviations: GVHD, graft-versus-host disease; -, negative effect; +, positive effect; 0, no significant effect | ||
| Outcome | ||
|---|---|---|
| Variable (Reference) | GVHD | Survival |
| Chronic phase status at time of SCT (59,62) | - | + |
| Serologic mismatch Class I or II (59,61,65) | + | 0 |
| Molecular mismatch (62,66,67) | + | - |
| Older age (59,62) | 0 | - |
| Transplant < 12 months from diagnosis (59,62) | 0 | + |
| CMV positivity (59,61) | 0 | - |
| T-cell depletion (59) | - | 0 |
| Abbreviations: GVHD, graft-versus-host disease; -, negative effect; +, positive effect; 0, no significant effect | ||
The approach to patients with CML who lack an HLA-identical sibling donor has become increasingly difficult. On one hand the availability of non-transplant strategies that can effectively achieve cytogenetic remissions, such as interferon and more recently STI571, offers the possibility of long-term disease control for some patients without allogeneic transplantation.68 On the other hand, the increasing size and number of volunteer donor registries as well as improvements in tissue typing and supportive care have improved the outcomes of unrelated donor transplantation. The current challenge for physicians and patients is how to incorporate these strategies in each individual case and how to decide who should undergo unrelated donor transplantation as primary therapy or as salvage therapy for their disease. Transplant and non-transplant options should be viewed as complementary strategies in the treatment of CML, the goal being to obtain complete cytogenetic remissions in as many patients as possible with the minimum toxicity necessary for each patient.
Donor Lymphocyte Infusions
DLI are an effective therapeutic strategy for patients with CML who have relapsed after allogeneic hematopoietic transplantation.69,70 DLI is limited by the occurrence of acute and chronic GVHD in up to 60% of patients, which has been associated with significant morbidity and a mortality of approximately 20%. Current strategies to improve the results of DLI in CML have concentrated on decreasing the risk of GVHD after infusion, and fall into one of the following categories:
Selective depletion of lymphocyte subsets.
Incremental doses of donor lymphocytes.
Gene-modified DLI
CD8-depleted donor lymphocyte infusions
Depletion of CD8+ cells from DLI has resulted in a reduction of the incidence of GVHD without compromising response.71 Remissions after CD8-depleted DLI are durable and the factors that predict outcome are similar to those after unmanipulated DLI and are summarized in Table 5.
Predictors of outcome post donor lymphocyte infusion (DLI) in chronic myelogenous leukemia (CML) relapse following allogeneic stem cell transplantation (SCT). Results compare presence vs absence of the variable in question.
| Study (Reference)/Variables | % Response | % GVHD | % Aplasia | |||
|---|---|---|---|---|---|---|
| Variable | P | A | P | A | P | A |
| Kolb(69) | ||||||
| CP at DLI | 80 | 2 | ||||
| No GVHD with BMT | 84 | 64 | ||||
| IFN treatment | 73 | 51 | 52 | 24 | ||
| Hematologic relapse | 68 | 45 | 49 | 12 | ||
| Collins(70) | ||||||
| CP at DLI | 81 | 44 | ||||
| No GVHD with BMT | 64 | 86 | 72 | 91 | ||
| Caucasian | 64 | 85 | 72 | 94 | ||
| IFN treatment | 74 | 81 | ||||
| Shimoni(71) | ||||||
| CP at DLI | 87 | 9 | ||||
| Time to relapse < 12 months | 23 | 85 | ||||
| Sibling donor | 40 | 100 | ||||
| Dazzi(74) | ||||||
| Bulk DLI | 67 | 91 | 44 | 10 | ||
| Abbreviations: P, presence; A, absence; CP, chronic phase; DLI, donor lymphocyte infusion; GVHD, graft-versus-host disease; BMT, bone marrow transplantation; IFN, interferon | ||||||
| Study (Reference)/Variables | % Response | % GVHD | % Aplasia | |||
|---|---|---|---|---|---|---|
| Variable | P | A | P | A | P | A |
| Kolb(69) | ||||||
| CP at DLI | 80 | 2 | ||||
| No GVHD with BMT | 84 | 64 | ||||
| IFN treatment | 73 | 51 | 52 | 24 | ||
| Hematologic relapse | 68 | 45 | 49 | 12 | ||
| Collins(70) | ||||||
| CP at DLI | 81 | 44 | ||||
| No GVHD with BMT | 64 | 86 | 72 | 91 | ||
| Caucasian | 64 | 85 | 72 | 94 | ||
| IFN treatment | 74 | 81 | ||||
| Shimoni(71) | ||||||
| CP at DLI | 87 | 9 | ||||
| Time to relapse < 12 months | 23 | 85 | ||||
| Sibling donor | 40 | 100 | ||||
| Dazzi(74) | ||||||
| Bulk DLI | 67 | 91 | 44 | 10 | ||
| Abbreviations: P, presence; A, absence; CP, chronic phase; DLI, donor lymphocyte infusion; GVHD, graft-versus-host disease; BMT, bone marrow transplantation; IFN, interferon | ||||||
At the University of Texas M.D. Anderson Cancer Center, 26 patients with CML who relapsed after an allogeneic transplant have been treated with CD8-depleted DLI. CD8+ depletion with immunomagnetic or panning techniques resulted in a mean depletion of 2.6 log of CD8+ lymphocytes. The median CD4+ cell dose infused with the first DLI was 39.8 × 106 cells/kg, and the median CD8+ dose was 0.3 × 106 cells/kg. Fourteen patients achieved complete cytogenetic and molecular remissions following DLI. The stage of the disease at the time of DLI was the most important prognostic factor for response. Thirteen of the 15 (87%) patients who were in cytogenetic or chronic phase relapse at the time of DLI responded. Actuarial rate was 8% (95 CI, 3-13%) for acute GVHD. Chronic GVHD occurred in 4 patients and was extensive in two patients. Actuarial risk for chronic GVHD was 22%.
The median follow-up of the 15 patients relapsing in non-transformed phases was 3.5 years (range, 1-6.5): 13 are still alive, 10 in complete remission; the estimated 5-year survival rate is 87%, and estimated 5-year disease-free survival rate 65% (Figure 3). This experience, as well as studies performed at the Dana-Farber Institute,(72) demonstrate that CD8-depleted DLI is effective in inducing remissions in post-transplant CML relapse, and reducing the risk of GVHD. These results are currently being tested in a prospective randomized trial.

Overall survival of CML patients receiving CD8-depleted donor lymphocyte infusions (DLI) for relapse after allograft according to disease status at the time of DLI.
Overall survival of CML patients receiving CD8-depleted donor lymphocyte infusions (DLI) for relapse after allograft according to disease status at the time of DLI.
Dose-escalated donor lymphocyte infusions
The incidence of GVHD increases with the number of cells infused, but there may be a different threshold for GVHD and graft-versus-leukemia (GVL). One approach is to administer escalating doses of donor lymphocytes starting with a relatively low dose and escalating the dose on subsequent infusions, infused in non-responders at predetermined intervals. Investigators at the Hammersmith Hospital have compared an escalating dose regimen, starting with 1 × 107 CD3+ cell/kg (1 × 106 for DLI from unrelated donors), to a single larger dose DLI (median dose 1.5 × 108 CD3+ cells/kg). The escalating dose regimen resulted in similar response rate but a lower incidence of acute and chronic GVHD, despite the infusion of an equivalent total number of lymphocytes with the multiple infusions.74
Transduction of donor lymphocytes with suicide genes
Transduction of donor cells with a suicide gene, such as the herpes simplex virus thymidine kinase, which confers sensitivity to ganciclovir, has also been explored as a means of increasing the safety of DLI. If the patient develops GVHD, it can be abrogated by ganciclovir treatment.75 In a recent update of this strategy Bonini et al reported a response rate of 60%; three patients developed ≥ grade 2 GVHD, of which two responded to ganciclovir therapy. However, in a multicenter study of TK-transduced DLI, Champlin et al reported only two responses in 14 patients with CML and no GVHD. This suggests that transduction techniques may affect the alloreactivity of the donor lymphocytes.76
Non-Ablative Stem Cell Transplantation
The anti-leukemia responses observed after DLIs in CML patients who relapsed after an allogeneic progenitor cell transplant represent direct evidence of the GVL effect mediated by lymphocytes. This observation also demonstrated that very effective GVL effects can be seen in CML without the need for the intense myeloablation provided by the preparative regimen. This finding paved the road for the concept of non-ablative stem cell transplantation (NST).77 NST has been proposed as a strategy that would allow the “harnessing” of the GVL effect in patients considered poor candidates for myeloablative therapies either because of their age or concurrent medical conditions.
CML represents an ideal candidate for therapy with NST for the following reasons: a) relatively stable natural history that will allow time for induction of a GVL effect; b) high response rate to immune-modulatory maneuvers (DLI, IFN); and c) lack of therapies that can change the natural course of the disease if failure of frontline therapy (usually IFN-based therapy) occurs.
CML patients also present particular challenges for NST due to their relatively conserved immune competence, which could increase the risk of rejection, and the presence of splenomegaly in some patients, which may affect engraftment.
Trials of non-ablative stem cell transplantation in CML are still immature and have only a small number of patients. At the M.D. Anderson Cancer Center, a total of 43 patients with CML have received either a true non-ablative stem cell transplant with fludarabine/idarubicin/cytarabine combinations or a reduced-intensity conditioning with melphalan/purine analog combinations. Patient characteristics are summarized in Table 6.
Characteristics of chronic myelogenous leukemia (CML) patients rndergoing non-ablative stem cell transplantation (Flag-Ida) or reduced intensity conditioning (fludarabine/melphalan) at M.D. Anderson Cancer Center.
| Parameter | Flag-Ida | Fludarabine/Melphalan |
|---|---|---|
| Number | 10 | 33 |
| Median age, (range) | 57 (42-72) | 52 (24-70) |
| Median time to BMT in months, (range) | 29 (13-11) | 29 (3-220) |
| Stage at BMT | ||
| first chronic | 5 | 8 |
| phase transformed | 5 | 25 |
| Donor type | ||
| sibling | 8 | 15 |
| unrelated | 2 | 18 |
| Comorbid conditions | ||
| age > 55 year | 6 | 13 |
| extensive prior therapy | 0 | 23 |
| infection/organ function/PS = 2 | 3 | 6 |
| none | 1 | 6 |
| Parameter | Flag-Ida | Fludarabine/Melphalan |
|---|---|---|
| Number | 10 | 33 |
| Median age, (range) | 57 (42-72) | 52 (24-70) |
| Median time to BMT in months, (range) | 29 (13-11) | 29 (3-220) |
| Stage at BMT | ||
| first chronic | 5 | 8 |
| phase transformed | 5 | 25 |
| Donor type | ||
| sibling | 8 | 15 |
| unrelated | 2 | 18 |
| Comorbid conditions | ||
| age > 55 year | 6 | 13 |
| extensive prior therapy | 0 | 23 |
| infection/organ function/PS = 2 | 3 | 6 |
| none | 1 | 6 |
Of the 10 patients who received fludarabine/idarubicin/cytarabine followed by infusion of donor progenitor cells, all had neutrophil and platelet recovery at a median of 13 days and 14 days respectively. All patients showed evidence of donor cell engraftment except the two recipients of unrelated donor cells. Bone marrow on day 30 revealed complete cytogenetic remission in four patients and major cytogenetic remission in another two patients. All five patients transplanted in advanced CML phases had cytogenetic progression during the first 6 months after transplant, and failed to respond to subsequent salvage therapy. Three of the five patients transplanted in chronic phase CML have relapsed. One patient achieved a subsequent complete cytogenetic remission with a donor lymphocyte infusion; the other two have failed to respond to this therapeutic maneuver. Survival and disease-free survival are depicted in Figure 4.

Current disease-free survival and overall survival for patients with chronic myelogenous leukemia (CML) undergoing non-ablative stem cell transplantation (Flag-Ida) at M.D. Anderson Cancer Center according to disease stage at transplant.
Current disease-free survival and overall survival for patients with chronic myelogenous leukemia (CML) undergoing non-ablative stem cell transplantation (Flag-Ida) at M.D. Anderson Cancer Center according to disease stage at transplant.
McSweeney et al, using 200 cGy of TBI, reported that four of eight patients with CML achieved complete cytogenetic remissions, with little toxicity, but graft failure rate was 20%, which has prompted the addition of fludarabine to the preparative regimen. More patients and longer follow-up will be needed to assess the true impact of non-ablative preparative regimens in the control of this disease.78
The initial results with fludarabine, idarubicin and ara-C combinations suggested that this strategy is not effective in CML in blast transformation and is not immuno-suppressive enough to allow engraftment of cells from unrelated or mismatched donors. Therefore, the development of reduced intensity conditioning, which may be more intense than truly non-ablative regimens but still better tolerated than conventional myeloablative regimens, was necessary. Melphalan in combination with purine analogs was thus developed as the new conditioning regimen in our studies.
Thirty-three patients with CML have undergone allogeneic transplants at M.D. Anderson Cancer Center after reduced intensity conditioning with fludarabine/melphalan combinations. More than half of these patients received bone marrow from unrelated donors, and only eight patients were in first chronic phase. Thirty-two of 33 patients achieved neutrophil recovery within a median of 14 days, and 24 achieved platelet transfusion independence at a median of 20.5 days. At the time of first marrow, the median percentage of donor cell engraftment as determined by cytogenetics or molecular techniques was 100% for recipients of sibling transplants (range, 80-100) and for recipients of unrelated donor bone marrow (range, 0-100). Only three recipients of unrelated donor bone marrow had less than 50% donor cells at the time of first marrow, one of whom became a full donor chimera over a period of 3 months. Other important transplant outcomes are summarized in Table 7 and depicted graphically in Figure 5.
Outcomes of non-ablative and reduced conditioning regimens for chronic myelogenous leukemia (CML) at M.D. Anderson Cancer Center.
| Parameter | Flag-Ida | Fludarabine/Melphalan |
|---|---|---|
| Number | 10 | 33 |
| Donor cell engraftment | 8 | 32 |
| Graft failure | 2 | 2 |
| Relapse | 6 | 8 |
| GVHD ≥ 2 | 1 | 14 |
| Responses after relapse | 2 | 1 |
| 2-yr survival | CP1:80% | CP1:63% |
| Other:20% | Other:33% | |
| 2-yr DFS | CP1:40% | CP1:63% |
| Other:0% | Other:30% |
| Parameter | Flag-Ida | Fludarabine/Melphalan |
|---|---|---|
| Number | 10 | 33 |
| Donor cell engraftment | 8 | 32 |
| Graft failure | 2 | 2 |
| Relapse | 6 | 8 |
| GVHD ≥ 2 | 1 | 14 |
| Responses after relapse | 2 | 1 |
| 2-yr survival | CP1:80% | CP1:63% |
| Other:20% | Other:33% | |
| 2-yr DFS | CP1:40% | CP1:63% |
| Other:0% | Other:30% |

Current disease-free survival of patients with chronic myelogenous leukemia (CML) undergoing reduced intensity conditioning with fludarabine/melphalan according to disease status at the time of transplant and donor type.
Current disease-free survival of patients with chronic myelogenous leukemia (CML) undergoing reduced intensity conditioning with fludarabine/melphalan according to disease status at the time of transplant and donor type.
In summary, NST and reduced intensity conditioning are feasible in CML and can result in long-term disease control. Their role in the therapy of CML remains to be established, in relation to the therapeutic alternatives of interferon-based therapy, STI571, and conventional myeloablative transplantation. Preliminary results suggest that NST should be further investigated as an alternative for patients in chronic phase ineligible for conventional transplant techniques, but that it may be ineffective in advanced phase CML.
Future Issues in Progenitor Cell Transplantation for CML
The main challenge for physicians and patients with CML is how to decide among existing therapeutic options. Current therapies should be viewed as complementary. Physicians and patients should develop treatment plans and algorithms from the time of diagnosis, deciding whether transplantation is appropriate as initial therapy or whether it will be used as salvage if initial non-transplant therapies fail to achieve complete cytogenetic remissions.
NST and reduced intensity conditioning have increased the pool of possible transplant candidates. However, GVHD, loss of donor cell engraftment, and disease recurrence are major causes of treatment failure. The incorporation of STI571 and/or IFN needs to be studied in the post-transplant setting as a means of reducing disease recurrence, particularly in patients who undergo progenitor cell transplantation in advanced phase CML.
IV. Interferon-α Plus Low-Dose Cytarabine and Other Promising Treatment Modalities for Chronic Myelogenous Leukemia
M.D. Anderson Cancer Center, Hematology Department, 1515 Holcombe Boulevard, Box 61, Houston TX 77030-4009
M.D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box 302, Houston TX 77030-4009
Investigational strategies in CML have aimed at suppression of Ph-positive CML cells (78,79) and more recently at suppression of the molecular consequences of the BCR-ABL abnormality or associated molecular events, as discussed earlier. Evidence supporting this approach include 1) the causal association between the BCR-ABL abnormality and development of CML, as demonstrated by the induction of CML-like disease by transfection of Ph-associated BCR-ABL molecular abnormalities in animal models80,81 ; 2) the suppression of CML disease achieved in preclinical models through interference with the BCR-ABL abnormalities82,83 ; and 3) the association between the achievement of minimal residual disease (complete hematologic response, cytogenetic response) and improved survival in CML.37,38,39,40,41 ,84
Large-scale studies with IFN-α, including those at M.D. Anderson, have reported a complete hematologic response (CHR) rate of 50-80% and a cytogenetic response rate of 20-60%.35,36,37,38,39,40,41 ,84,85 Patients achieving a major cytogenetic response have the greatest survival advantage; hence, strategies to improve this rate are warranted.
IFN-α and Low-Dose Cytarabine in Chronic Phase CML
The rationale behind the combination of IFN-α and low-dose cytarabine was based on 1) the selective in-vitro suppression of CML cells with cytarabine86 ; 2) the clinical experience with single-agent low-dose cytarabine showing cytogenetic responses in seven of nine patients87,88 ; and 3) the positive results from pilot studies in late chronic phase CML, which established the safety and efficacy of the combination.89
IFN-α and daily low-dose cytarabine in single-arm trials90,91,92,93
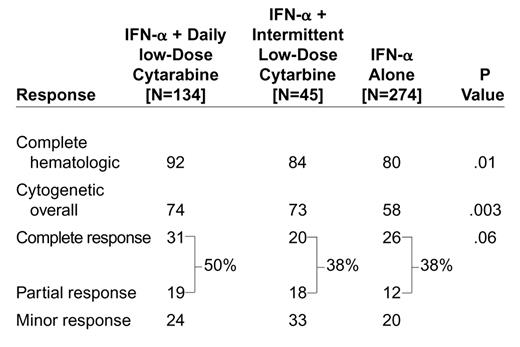
Experience with different schedules of the combination of IFN-α and low-dose ara-C have been reported in several single-arm studies in CML by the M.D. Anderson Cancer Center group90 (Tables8 and 9), Arthur et al from Australia,91 Thaler et al from Austria,92 and Silver et al for the CALGB Study.93 These studies demonstrated trends for higher response rates with the combination compared with IFN-α alone (Table 10). Table 11 computes the cumulative rates of CHR and cytogenetic responses reported from nine studies of IFN-α alone, as well as seven studies of IFN-α plus low-dose ara-C. In the MD Anderson studies, daily low-dose ara-C was more effective than intermittent low-dose ara-C (Table 8). Intermittent low-dose ara-C (20mg/m2 daily × 10 every months) may be associated with significant gastrointestinal complications (mucositis, diarrhea), and with frequent treatment interruptions due to myelosuppression.
Percent response to interferon-α (IFN-α) plus daily ara-C, IFN-α plus intermittent ara-C, and IFN-α alone.90
Prognosis by patient risk group with interferon-α (IFN-α) plus daily low-dose cytarabine therapy.
| % Cytogenetic Response | |||||
|---|---|---|---|---|---|
| Risk Group | No. | % CHR | Any | Major | CR |
| Good | 61 | 95 | 82 | 61 | 43 |
| Intermediate | 37 | 95 | 73 | 49 | 24 |
| Poor | 17 | 82 | 53 | 29 | 18 |
| P value | .17 | .05 | .045 | .06 | |
| Abbreviations: CHR, complete hematologic response | |||||
| % Cytogenetic Response | |||||
|---|---|---|---|---|---|
| Risk Group | No. | % CHR | Any | Major | CR |
| Good | 61 | 95 | 82 | 61 | 43 |
| Intermediate | 37 | 95 | 73 | 49 | 24 |
| Poor | 17 | 82 | 53 | 29 | 18 |
| P value | .17 | .05 | .045 | .06 | |
| Abbreviations: CHR, complete hematologic response | |||||
Single-arm studies of interferon-α (IFN-α) plus low-dose cytarbine.
Cumulative data on response with IFN-α with and without ara-C in CML
| Parameter | IFN-α | IFN-α + low-dose ara-C |
|---|---|---|
| No. treated | 1632 | 700 |
| No. studies | 9 | 6 |
| %CHR | 60 | 72 |
| % cytogenetic response | ||
| major | 22 | 42 |
| complete | 14 | 24 |
| Parameter | IFN-α | IFN-α + low-dose ara-C |
|---|---|---|
| No. treated | 1632 | 700 |
| No. studies | 9 | 6 |
| %CHR | 60 | 72 |
| % cytogenetic response | ||
| major | 22 | 42 |
| complete | 14 | 24 |
Randomized trials of IFN-α and low-dose cytarabine versus IFN-α alone
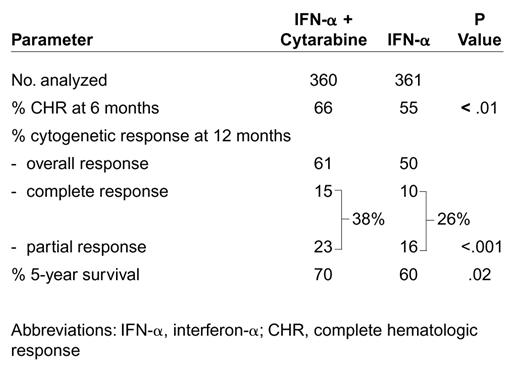
In a French multicenter trial in CML, Guilhot et al randomized patients to IFN-α 5 MU/m2 daily alone (N = 361) or IFN-α plus low-dose cytarabine 20 mg/m2 daily for 10 days every month (N = 360).94 IFN-α plus ara-C was associated with a significantly higher CHR rate at 6 months (66% versus 55%, p <.01), and a higher cytogenetic response rate at 12 months, overall (61% versus 50%) and major (38% versus 26%, p <.0.01) (Table 12). With a median follow-up of 43 months, IFN-α and cytarabine was associated with a significantly better survival (5-year rate 70% versus 60%; p =.02), which was confirmed by multivariate analysis. A landmark analysis by cytogenetic response at 2 years showed an association of cytogenetic response with survival: the 7-year survival rate was 85% for patients achieving complete or partial cytogenetic response, 62% for those with minor cytogenetic response, and 25% for others. Cytogenetic response, whether obtained with IFN-α alone or with IFN-α plus ara-C, was associated with significant prolongation of survival. These results continued to be confirmed with longer follow-up.95
The Italian Cooperative Study Group on CML (ICSG-CML) conducted a randomized trial similar to the French study. The ICSG-CML study used similar schedules of IFN-α plus cytarabine versus IFN-α. Five hundred forty evaluable patients were randomized; 275 received IFN-α and cytarabine, and 265 received IFN-α alone. In an update of the study in 1999, the mean observation time was 24 months. The combination of IFN-α plus ara-C was demonstrating superior results.96 At 6 months, the CHR rates were 87% versus 80%. IFN-α plus ara-C was associated with higher cytogenetic response rates (48% versus 44%), and with higher rates of major cytogenetic response (28% versus 19%, p =.01), and of complete cytogenetic response (14% versus 7%) compared with IFN-α alone. The 3-year survival rate was also higher with IFN-α plus ara-C compared with IFN-α alone (85% versus 80%, p =.03). However, these results need to be confirmed with longer follow-up and more mature analysis before a final assessment of the potential benefit of adding ara-C can be made (S. Tura, personal communication).
Homoharringtonine
Homoharringtonine (HHT) is a semi-synthetic plant alkaloid derived from the Cephalotaxus fortuneii tree. Initial studies from China suggested activity of HHT in acute myelogenous leukemia (AML) and acute promyelocytic leukemia. In fact, many frontline studies in AML use the combination of HHT and cytarabine with or without anthracyclines.
Phase I-II studies in the United States confirmed modest activity of HHT in AML and myelodysplastic syndrome. However, at HHT doses of 5 to 9 mg/m2 daily for 5 to 7 days, severe cardiovascular and hypotensive complications occurred, discouraging further investigations of HHT. At M.D. Anderson Cancer Center, a lower-dose longer exposure schedule of HHT (2.5 mg/m2 by continuous infusion daily for 14 to 21 days) reduced significantly the cardiovascular problems. HHT alone (2.5 mg/m2 daily × 14 days for induction, then × 7 days every month for maintenance)97 was then compared to HHT + ara-C (HHT 2.5 mg/m2 daily × 5 and ara-C 15 mg/m2 daily × 5, every month) in late chronic phase CML. Among 173 patients treated, the CHR and cytogenetic response rates were identical with either regimen (Table 13). However, survival was significantly longer with HHT + ara-C, even after accounting for risk groups and by multivariate analysis (Kantarjian, submitted).
Results with homoharringtonine (HHT) alone or with cytosine arabinoside (ara-C) in late chronic phase chronic myelogenous leukemia (CML) (p =.02).
| Parameter | HHT | HHT plus ara-C |
|---|---|---|
| No. treated | 73 | 100 |
| %CHR | 67 | 72 |
| % cytogenetic response | 30 | 32 |
| - major | 15 | 15 |
| - complete | 5 | 5 |
| % 4-year survival | 38 | 58 |
| Abbreviations: CHR, complete hematologic response | ||
| Parameter | HHT | HHT plus ara-C |
|---|---|---|
| No. treated | 73 | 100 |
| %CHR | 67 | 72 |
| % cytogenetic response | 30 | 32 |
| - major | 15 | 15 |
| - complete | 5 | 5 |
| % 4-year survival | 38 | 58 |
| Abbreviations: CHR, complete hematologic response | ||
In subsequent studies, HHT was investigated in early chronic phase CML. HHT was then given for six cycles as remission induction followed by IFN-α maintenance. CHR rate was 92% and cytogenetic response rate was 68%.98 Results after 6 months appeared to favor HHT over IFN-α in relation to response, suggesting that HHT may be a valuable addition to our future frontline therapies, which could include IFN-α, ara-C, STI571 and others. HHT is certainly superior to low-dose ara-C alone, with which one could not achieve the impressive results of HHT in patients in late chronic phase CML who failed IFN-α therapy.
After establishing the baseline efficacy of single-agent HHT for 6 months in frontline therapy of CML (to compare it with IFN-α), combination studies of HHT, IFN-α and ara-C, and of HHT + IFN-α in older patients were developed. So far, about 100 patients in early chronic phase have received the triple regimen. The CHR rate was 98%; with a median follow-up 11 months, the cytogenetic response rate was 77% (major 43%). Because of the short follow-up, the 6-month results of HHT + IFN-α + ara-C, compared with IFN-α + ara-C, or IFN-α alone were evaluated. These show improved cytogenetic response rates (Table 14). In addition lower doses of IFN-α (median 2MU daily), ara-C (median 5 mg Tiw), and HHT (median 2 days/month) were required to achieve the results. Anemia appeared to be a significant cumulative side effect, which required dose reduction and could be improved with weekly erythropoietin injections.99 Similar encouraging results were obtained with HHT + IFN-α, mostly in older patients.100
Six-month response rates with homoharringtonine (HHT) + cytosine arabinoside (ara-C) + interferon-α (IFN-α).
| 6-Month Response | HHT + IFN-α + Ara-C | IFN-α + Ara-C | IFN-α |
|---|---|---|---|
| No. patients | 42 | 148 | 274 |
| % CHR | 98 | 70 | 67 |
| % cytogenetic response | |||
| - major response | 67 | 49 | 39 |
| - complete response | 33 | 18 | 12 |
| Abbreviations: CHR, complete hematologic response | |||
| 6-Month Response | HHT + IFN-α + Ara-C | IFN-α + Ara-C | IFN-α |
|---|---|---|---|
| No. patients | 42 | 148 | 274 |
| % CHR | 98 | 70 | 67 |
| % cytogenetic response | |||
| - major response | 67 | 49 | 39 |
| - complete response | 33 | 18 | 12 |
| Abbreviations: CHR, complete hematologic response | |||
Ernst et al also combined HHT + ara-C in early and late chronic phase CML. Among 44 patients treated, 41 (93%) achieved CHR, including 14/14 (100%) receiving initial therapy. Among 14 patients treated with HHT + ara-C as initial therapy, 11 (84%) had a major cytogenetic response. In their studies, the CHR and cytogenetic response rates were higher than those reported in our studies, which may be due to the difference in treatment schedules or study group characteristics.101
We are currently investigating alternate routes of HHT delivery (e.g. subcutaneous or oral schedules), and possible modifications to the HHT structure, which could improve the efficacy: toxicity profile of HHT (Jean-Pierre Robin, personal communication). This could lead to investigation of HHT as an agent for treatment of other hematologic cancers such as myelodysplastic syndrome, AML maintenance, acute promyelocytic leukemia or others.
5-Aza-2′-Deoxycytidine
General and site-specific DNA methylation is characteristic of disease progression and resistance in many tumors, including solid cancers as well as hematologic malignancies such as myelodysplastic syndrome, acute leukemia, and CML. There is now major interest in strategies that suppress DNA methylation as a method to treat cancers or even non-malignant conditions such as sickle cell disease and thalassemia. 5-azacytidine and 5-aza-2′-deoxyazacytidine (decitabine) are cytidine analogues capable of inducing hypomethylation through inhibition of DNA methyltransferase enzyme. Both agents have already demonstrated significant activity in myelodysplastic syndrome, which is thought to be partly due to inhibition of methylation of p15INK4b. In CML, disease progression is associated with hypermethylation of the Pa promoter region within BCR-ABL.102,103 This led to our interest in using decitabine in CML. The initial studies used decitabine at 100 mg/m2 over 6 hours every 12 hours for 10 doses (5 days) every 4-8 weeks (1000 mg/m2/course), based on the previous European experience. This was associated with delayed myelosuppression and dose reductions of 50%, i.e. 50 mg/m2 per dose (500 mg/m2/course). Decitabine produced response rates of 25% in blastic phase and 53% in accelerated phase disease.104 Compared with intensive chemotherapy as initial therapy for CML blastic phase, decitabine was associated with significantly better survival among patients 50 years or older (p <.01). By multivariate analysis, decitabine therapy remained an independent favorable prognostic factor for survival (p =.047) after accounting for pretreatment prognostic factors.105 We are currently investigating lower dose schedules of decitabine (e.g. 5 to 10 mg/m2 daily for 10 to 20 days) to target hypomethylation as the molecular endpoint and correlate it with response. This may provide a less myelosuppressive, and perhaps equally effective regimen. Investigations of decitabine in combination with busulfan and cyclophosphamide as part of a preparative regimen for allogeneic stem cell transplantation and as salvage therapy with stem cell rescue after relapse from allogeneic transplant are also in progress.106
Polyethylene Glycol (PEG) Interferon
Polyethylene glycol (PEG) interferon is a modified IFN-α molecule that is covalently attached to polyethylene glycol. PEG interferon has a longer half-life than the parent compound and is given once weekly instead of daily. In a phase I study, 21 patients with CML in chronic phase were treated with escalating doses of PEG interferon (Intron). The starting dose of PEG-Intron was 0.75 μg/kg weekly. The presumed equivalent dose of regular IFN-α 3 to 5 MU/m2 daily (i.e. 21 to 35 MU/m2 weekly) is a weekly dose of PEG Intron 1.2 to 1.5 μg/kg. The maximum tolerated dose in the study was 7.5 μg/kg weekly, and the dose chosen for phase II studies of PEG-Intron was 6 μg/kg weekly (presumably equivalent to regular IFN-α 180 MU weekly = 15 MU/m2 daily). Dose-limiting toxicities were neurotoxicity, thrombocytopenia, fatigue, and liver function abnormalities, similar to IFN-α, but occurring at higher doses than with IFN-α. In addition to a better side-effect profile of PEG interferon, 9/27 patients improved their responses, including three complete and three partial cytogenetic responses. All six patients intolerant to IFN-α were able to receive Peg-Intron. Preliminary results with PEG interferon are promising since it appears to be easier to deliver (once weekly), less toxic, and possibly more effective than IFN-α. Current studies are investigating the two different formulations of PEG interferon, PEG-Intron and Pegasys (PEG-Roferon) alone and in combination with ara-C in early chronic phase CML. PEG-Roferon has a larger molecular weight and may have less CNS penetration but more hepatic uptake. It is likely that PEG interferon preparations will replace IFN-α in future standard of care because of the easier route of delivery, if the favorable efficacy:toxicity profile is confirmed with longer follow-up.
V. STI571 in Chronic Myelogenous Leukemia
Hagop M. Kantarjian, M.D., and Moshe Talpaz, M.D.
STI571 represents a major therapeutic advance in the management of CML. The experience with STI571 provides the most dramatic example of how understanding disease biology and pathophysiology can result in extremely successful molecular-targeted approaches. CML is an ideal disease for such advances, since the molecular target is unique, highly expressed in most patients, and disease specific. STI571 may provide a model applicable to many other tumors in the future.
General Overview of BCR-ABL Signal Transduction Inhibitors
As discussed previously, several naturally occurring compounds manifest inhibitory activity against protein tyrosine kinases, including the isoflavinoid genistein, herbimycin A (an antibiotic), the flavinoid quercetin, and erbstatin.108,109 Later, synthetic compounds known as tyrphostins were rationally designed to complete with ATP or substrate for the binding site in the catalytic center of the kinase.18,110,111 These were the first compounds to display specificity for tyrosine kinases.112 Scientists at Novartis identified kinase inhibitors by screening a large library of compounds. Optimization of a lead compound against specific targets led to the development of STI571.
Preclinical Experience with STI571
STI571 inhibits the ABL tyrosine kinases at very low concentrations (0.025 micromolar), including p210BCR-ABL, p185BCR-ABL, V-ABL, and c-ABL. In addition, STI571 inhibits the c-kit and platelet-derived growth factor receptor (PDGFR) tyrosine kinases at similar concentrations.21,23,113
This is of interest for several reasons:
The submicromolar concentrations were easily achievable in the clinical trials. Once the STI571 dose in phase I studies reached 300 mg orally daily, serum levels of STI571 of ≥ 1 micromolar were achieved and correlated with high response rates.
STI571 has activity in both p210 and p190 Bcr-Abl-positive leukemias.
The therapeutic activity of STI571 may extend beyond Ph-positive leukemias to c-kit and PDGFR-positive disorders including AML, myeloproliferative disorders, prostate cancer, brain tumors, sarcomas and others.
Preclinical studies have confirmed the selective inhibitory effects of STI571 against p210BCR-ABL and p190BCR-ABL,20,21 with minimal inhibition of normal hematopoiesis. Inhibitory activity results in apoptosis. Efficacy was also demonstrated in syngeneic or nude mice injected with BCR-ABL-expressing cells and then treated with STI571.23,114
Animal toxicology studies with oral STI571 demonstrated progressive hepatic toxicity at the highest dose of 100 mg/kg. Other toxicities were vomiting, diarrhea, and myelosuppression (anemia, neutropenia).
Update of the Clinical Experience with STI571 in CML
STI571 has undergone relatively rapid evaluations in phase I and phase II pivotal trials for the purpose of FDA approval. The pivotal trials targeted the three phases of CML, chronic, accelerated and blastic. The studies opened in December 1999, and patient accrual was completed by May 2000.
The phase I dose escalation study used STI571 at a starting daily dose of 25 mg orally and reached a highest dose of 1000 mg. Common side effects were nausea, skin rashes, muscle cramps and bone aches; these were mostly mild and responsive to supportive care. Myelosuppression with anemia, granulocytopenia and thrombocytopenia were also observed. Hepatotoxicity was rare. At the higher doses, edema (periorbital, leg) and fluid retention were more prominent, but there were no clear-cut maximally tolerated dose (MTD) or dose-limiting toxicity (DLT). The phase I study continues with lower doses of STI571 in combination with low-dose ara-C.
The phase I study included both chronic and blastic phase patients. The results in chronic phase CML patients who had failed IFN-α therapy were recently updated.115 At daily oral STI571 doses of 300 mg or more, the CHR rate was 100% and cytogenetic responses were observed 53% of patients with complete cytogenetic remissions in 13%. Most of these responses are maintained with ongoing therapy.
The results of the phase I study in blastic phase were also recently reported at ASCO.116 A total of 51 patients were treated with STI571 at doses of 300 to 800 mg orally daily, and 48 were evaluable. Their median age was 53 years (range 26 to 76 years); 33 had myeloid blastic phase, seven had lymphoid blastic phase, and eight had Ph-positive ALL. Responses were defined as complete response (classical definition), complete response without recovery of peripheral counts (i.e. marrow CR), and partial responses (marrow blasts reduction to < 15%). The results are summarized in Table 15. Most responses were transient, lasting for a median of 3 months; few complete responses from myeloid blastic phase are ongoing after one year. Three patients had complete cytogenetic responses.
Results of STI571 in chronic myelogenous leukemia (CML) blastic phase.
| No. (%) | |||
|---|---|---|---|
| Parameter | Myeloid | Lymphoid and Ph-positive ALL | Total |
| No. treated | 33 | 15 | 48 |
| Complete response | 4 (12) | 3 (20) | 7 (15) |
| Marrow complete response | 5 (15) | 6 (40) | 11 (23) |
| Partial response | 15 (45) | 2 (13) | 17 (35) |
| No. (%) | |||
|---|---|---|---|
| Parameter | Myeloid | Lymphoid and Ph-positive ALL | Total |
| No. treated | 33 | 15 | 48 |
| Complete response | 4 (12) | 3 (20) | 7 (15) |
| Marrow complete response | 5 (15) | 6 (40) | 11 (23) |
| Partial response | 15 (45) | 2 (13) | 17 (35) |
The pivotal trials have accrued over 500 patients in chronic phase and over 200 patients in accelerated phase. Updated results of the STI571 studies will be presented at the ASH meeting.
STI571 is currently being compared to IFN-α plus ara-C in a randomized trial of newly-diagnosed patients. Combinations of STI571 with low-dose ara-C and with IFN-α are in progress or planned. For CML in blastic phase and Ph-positive ALL, combinations of STI571 with standard anti-leukemic agents warrant study. Other potential applications of STI571 to treat minimal residual disease (post SCT or IFN-α), for in vitro purging, and in combinations with other agents are under evaluation. More time is needed to evaluate the drug's effect on survival and potential cure of CML. Finally, ongoing studies are assessing mechanisms of drug resistance.
VI. Summary and Future Directions
Major breakthroughs have recently occurred in understanding the pathophysiology of CML, and in its treatment. Of the four main therapeutic breakthroughs—allogeneic SCT, DLI, IFN-α regimens, and STI571—STI571 may have the most significant impact on patient prognosis.
It is clear that many of our current practical and investigational strategies in CML will evolve around STI571-based regimens. Rapid evaluations of STI571 alone and in combination are needed in newly diagnosed CML patients, in advanced phase CML and in Ph-positive ALL. Other molecularly targeted strategies (e.g. FTIs) and immunomodulatory approaches (cellular, vaccines) need to be evaluated. Patients failing STI571 and who have received or are not candidates for SCT or IFN-a regimens need to undergo investigational approaches designed specifically for STI571 failure, aimed at reversing the mechanisms of resistance or at using alternate strategies (e.g. HHT, decitabine). Finally the role and timing of allogeneic SCT and IFN-α regimens need to be re-evaluated frequently as the STI571 results continue to mature.
