

Abstract
Cold-reactive autoimmune hemolytic anemia (AIHA) is rare among the hemolytic anemias. It results when 1 of a variety of processes causes the generation of immunoglobulin M (IgM) autoantibodies against endogenous erythrocytes, resulting in complement activation and predominantly intravascular hemolysis. Cold AIHA is typically a primary lymphoproliferative disorder with marrow B-cell clones producing pathogenic IgM. More rarely, secondary cold AIHA (cAIHA) can develop from malignancy, infection, or other autoimmune disorders. However, in children cAIHA is typically post infection, mild, and self-limited. Symptoms include a sequelae of anemia, fatigue, and acrocyanosis. The severity of disease is variable and highly dependent on the thermal binding range of the autoantibody. In adults, treatment has most commonly focused on reducing antibody production with rituximab-based regimens. The addition of cytotoxic agents to rituximab improves response rates, but at the expense of tolerability. Recent insights into the cause of cold agglutinin disease as a clonal disorder driven by complement form the basis of newer therapeutic options. While rituximab-based regimens are still the mainstay of therapy, options have now expanded to include complement-directed treatments and other B-cell-directed or plasma-cell-directed therapies.
Learning Objectives
Understand the role of novel therapeutics in the treatment of primary cold agglutinin disease
Compare the management of adult and pediatric cold autoimmune hemolytic anemia
CLINICAL CASE 1
A 65-year-old woman is admitted for anemia identified during a workup for fatigue. Her hemoglobin level is 6.5 g/dL, and she is sent to the hospital for admission. She reports purple discoloration of her fingers during the winter. Her exam demonstrates pallor, scleral icterus, and jaundice. She has a positive polyspecific direct antiglobulin test (DAT), negative immunoglobulin G (IgG) and positive C3d, undetectable haptoglobin, lactate dehydrogenase (LDH) of 620 U/L, unconjugated hyperbilirubinemia at 4.8 mol/L, and an elevated absolute reticulocyte count (ARC) at 240 × 109/L. Her C3 and C4 are undetectable, and cold agglutinin titer is 1024 at 4 °C. Bone marrow (BM) biopsy does not identify malignancy but is consistent with cold agglutinin-associated lymphoproliferative BM disease, with nodular B-cell aggregates, the absence of paratrabecular growth, and the fibrosis and lymphoplasmacytoid cells seen in lymphoplasmacytic lymphoma. Serum protein electrophoresis demonstrates monoclonal IgMκ. The sample is negative for MYD88 L265P mutation. Flow cytometry on her BM sample demonstrates a ratio of κ/λ-positive B cells of 6. She is diagnosed with primary cold agglutinin disease (CAD). She receives a warmed erythrocyte transfusion and is started on rituximab and bendamustine. As a bridge pending B-cell depletion, she starts weekly sutimlimab infusions, which are spaced to every 2 weeks after the first 2 doses. Her hemolysis resolves and she remains in remission at 3 months. Her sutimlimab is discontinued without any further hemolytic anemia.
CLINICAL CASE 2
An 11-year-old boy is admitted for severe anemia and hyperbilirubinemia. Ten days prior, he had nasal congestion, rhinorrhea, cough, and low-grade fever. His symptoms progressed to include headache, dizziness, pallor, worsening fatigue, dark urine, and yellowing of his eyes. His pediatrician referred him to the emergency room. On exam, he is tachycardic with a systolic ejection murmur. He has diffuse pallor, jaundice, and scleral icterus, as well as mild hepatosplenomegaly. His labs show a white blood cell count of 344 × 109/L, a hemoglobin level of 5.4 g/dL, a mean corpuscular volume of 104.3 fL, and a platelet count of 344 × 109/L. He has an inappropriately low reticulocyte count (2.8%; ARC, 38 000). The peripheral smear shows normocytic anemia with spherocytes, clumped cells, and an absence of schistocytes. DAT IgG is negative and DAT C3 2 is positive, with cold autoantibody identified. He has a mild transaminitis, unconjugated hyperbilirubinemia, undetectable haptoglobin, and elevated LDH. He is transfused at presentation using a blood warmer and is started empirically on a 5-day course of azithromycin. He is found to be mycoplasma IgM and IgG positive. He requires a second transfusion 2 days later for a hemoglobin level of 6.4 g/dL. He remains hospitalized for 5 days with his room kept warm and is advised against cold showers and drinking cold beverages. At the time of discharge, his hemoglobin level is stable at 8.1 g/dL, his ARC is 152 000 (5.7%), and his hemoglobinuria and jaundice have resolved. His hemoglobin normalizes within 2 weeks during outpatient follow-up.
Introduction
Cold autoimmune hemolytic anemia (cAIHA) is caused by IgM autoantibodies whose κ light chains bind erythrocyte I (or i) antigens at temperatures below 37 °C.1 IgM-bound red blood cells (RBCs) agglutinate and activate complement. C3b on the RBC surface triggers phagocytosis via the hepatic reticuloendothelial system. Terminal complement is activated in the vasculature, leading to intravascular hemolysis.2
Cold AIHA is commonly primary but can be secondary to another disorder. In adults, CAD is a clonal disorder driven by low-grade B-cell proliferation in the absence of an overt malignancy. Proliferating B cells produce IgM, which drives hemolysis through complement activation (Visual Abstract).2 CAD is more common than cold agglutinin syndrome (CAS), which is defined as cold hemolytic anemia arising secondary to another disorder such as autoimmune disease, infection, or malignancy.3
Diagnostic workup
The recommended workup for cAIHA is outlined in Table 1. The patient history and physical exam require attention to signs of malignancy or infectious etiology, including evaluation for lymphadenopathy and hepatosplenomegaly. Evidence of acrocyanosis is important in both diagnosis and for guiding treatment choices. Patients may report worse symptoms in colder temperatures or exacerbations during illnesses.4
Direct antibody testing
The initial laboratory workup demonstrates hemolysis with an elevated reticulocyte count and positive hemolytic markers. A positive DAT confirms immune-mediated hemolysis. The DAT is typically strongly C3d positive but may also be weakly IgG positive for reasons not fully understood. IgM readily activates the classical complement pathway on the erythrocyte surface. Upon warming, the antibody detaches from the RBC surface before it can be detected in the DAT assay, but bound C3b remains. C3b-bound erythrocytes are phagocytosed by the hepatic reticuloendothelial system (extravascular hemolysis). For cells that survive phagocytosis, surface C3b is degraded into less active complement components, including C3d, which is reported in clinical DAT results. Intravascular hemolysis occurs via terminal complement activation with lysis from the action of the C5b-C9 membrane attack complex on the RBC surface (Visual Abstract).5 The classic teaching is that cAIHA is primarily intravascular; however, extravascular hemolysis in the liver predominates, especially in stable disease. Baseline activity of the regulatory proteins CD55 and CD59 impedes complement activation on the RBC surface, and intravascular hemolysis plays a larger role in disease exacerbation or severe disease.5
Performing a DAT and antibody identification can be technically challenging. With a full understanding of the clinical scenario, transfusion medicine may be able to optimize testing and also provide hematology insights into DAT strength or the behavior of antibodies in the sample. Collaboration with transfusion medicine in the management of cAIHA is also critical for treatments such as transfusion or plasmapheresis.
Other laboratory features
The mean corpuscular volume is elevated, reflecting reticulocytosis, or falsely elevated due to RBC agglutination.6 RBC agglutination is apparent on peripheral smear (Figure 1). As with the patient in Clinical Case 1, complement levels are typically low due to consumption.7 The cold agglutinin titer is at least 64 in CAD and often substantially higher. Cold agglutinin thermal properties have implications for the clinical course: a higher thermal amplitude, which is the highest temperature at which the cold agglutinin binds, equates to antibodies binding at a wider range of temperatures and more severe disease. This testing is technically difficult, requiring the sample to be maintained at 37 °C to 40 °C until the serum has been removed.4
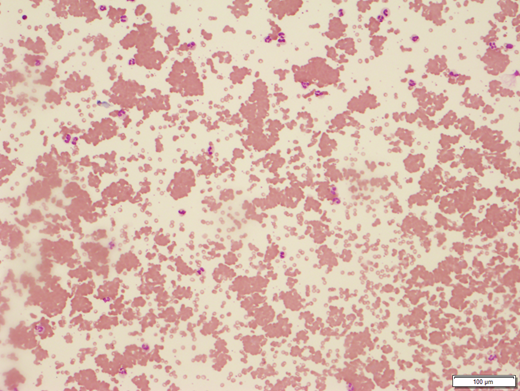
Peripheral blood smear from patient with cold agglutinin disease showing red blood cell clumping (100 × ). Image courtesy of Dr Tarek M. Elghetany.
Peripheral blood smear from patient with cold agglutinin disease showing red blood cell clumping (100 × ). Image courtesy of Dr Tarek M. Elghetany.
Patients should be evaluated for infection, the presence of an overt malignancy, or systemic autoimmunity. CAS refers to cold antibody–mediated hemolytic anemia that has developed secondary to another condition. CAD refers to primary cAIHA driven by the low-grade lymphoproliferation of clonal B cells producing IgM. Serum protein electrophoresis confirms a CAD diagnosis and typically shows monoclonal IgMκ. Required for adult patients in order to distinguish CAS and CAD, BM assessment demonstrates clonal lymphoproliferative infiltration, and immunophenotyping shows an abnormal B-cell clone with a B-lymphocyte κ/λ ratio greater than 3.5.8 A computed tomographic scan of the chest, abdomen, and pelvis is necessary to rule out underlying malignancy, particularly lymphoma.
Considerations for treatment
CAD management is based on the degree of anemia and symptomatology. Those with mild or compensated anemia may not need treatment. Disease exacerbations occur in infection, surgery, or cold exposure and may necessitate intervention.1,9 In a study of 232 CAD patients, 36% had mild (hemoglobin level >10) or fully compensated anemia.1 Another 37% had moderate anemia (hemoglobin 8-10 g/dL), and 27% had severe anemia with hemoglobin levels lower than 8.1 Some patients are transfusion dependent, with an estimated 40% of patients having been transfused.1,7,10 Those living in colder climates experience cold- induced ischemic symptoms, including cyanosis of areas farther from the body core such as the tips of the ears, the nose, and the digits. While treatment for moderate to severe anemia or bothersome acral symptoms is accepted, fatigue as an indication for therapy is less clear.11 Fatigue is a prominent complaint for many patients and thought to correlate with complement activity.5 Independent of anemia severity, CAD carries an increased risk for thromboembolic events, including stroke, myocardial infarction, and deep venous thrombosis, which should be considered in making therapy decisions.1
Supportive management
Patients should avoid cold temperatures, with attention to keeping acral areas warm.10,12 For symptomatic anemia, transfusion can be given using an in-line blood warmer. Without warming, hemolysis is exacerbated, leading to line occlusion during the transfusion.12
Plasmapheresis may be indicated for patients with severe, symptomatic anemia requiring rapid intervention. A 1 to 1.5 times plasma volume exchange decreases IgM and improves anemia.13 For children, plasmapheresis is used as a temporizing measure pending spontaneous disease resolution. While in adults with CAD, plasmapheresis should be followed by more durable therapy.14
Therapy to avoid
In warm AIHA, opsonized RBCs are destroyed by splenic macrophages. In cAIHA, C3b-coated RBCs are primarily phagocytosed by the hepatic reticuloendothelial cells; therefore, splenectomy is not indicated in most CAD cases.14
Corticosteroids, the mainstay of treatment in warm AIHA, are ineffective in cAIHA and should be avoided.17 Despite a lack of supporting data and an unfavorable side effect profile, corticosteroids are widely employed.1,7,18 Intravenous Ig is ineffective in AIHA, in contrast to its use in other immune cytopenias.19 Other immune suppressive agents are of limited utility.17
Pharmacologic management
B-cell directed therapy
Therapies for CAD are summarized in Table 2, and a proposed treatment algorithm is presented in Figure 2. Rituximab is the mainstay of treatment for CAD. It is tolerated well and induces responses in 45% to 60% of patients. Complete responses are rare.20,21 The median time to response to rituximab is approximately 1.5 to 3 months, with a median remission duration of approximately 6 months.20,21 Most patients experience relapse upon B-cell repopulation, typically within 1 year.21
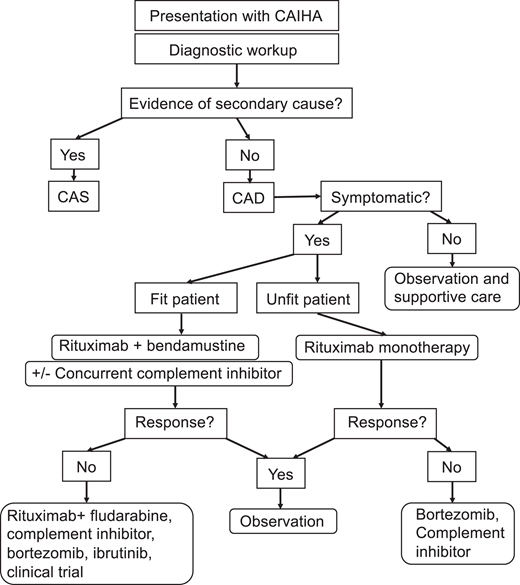
Treatment algorithm for cold agglutinin disease. Adapted with permission from S. Berentsen.17
Treatment algorithm for cold agglutinin disease. Adapted with permission from S. Berentsen.17
Given its moderate efficacy and high relapse rate with monotherapy, rituximab is used in combination with the cytotoxic agents fludarabine and bendamustine. Rituximab with fludarabine has superior efficacy over rituximab alone and may induce responses in those refractory to rituximab monotherapy.22 However, fludarabine has higher toxicity; in a prospective uncontrolled trial, 57% of patients developed an infection, and 41% had grade 3 to 4 hematologic toxicity (14% with grade 4 neutropenia).22 Rituximab with bendamustine has an approximately 70% overall response rate with a more favorable side effect profile.
For those with comorbidities, rituximab monotherapy is the recommended first-line therapy, while for fit individuals likely to tolerate cytotoxic therapy, rituximab with bendamustine is the treatment of choice.17 For those who fail initial treatment or have relapsing symptoms, second-line therapies include complement inhibitors, bortezomib, or, for those well enough to tolerate it, ibrutinib or rituximab with fludarabine.17
Complement-directed therapy
The intravascular component of CAD is driven by terminal complement activation on the RBC surface (Visual Abstract). Eculizumab, an anti-CD5 monoclonal antibody inhibiting terminal complement, is used in other complement-driven conditions, including paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome.25,26 The DECADE trial was an open-label, prospective nonrandomized phase 2 trial of eculizumab in CAD patients.27 Eculizumab showed an overall response rate of 54%, with response defined as a decrease in LDH greater than or equal to 250 U/L. The median increase in hemoglobin level was modest, at less than 1 g/dL over the 26-week study period.28 Patients with a narrower thermal amplitude (indicating milder disease) had a better response to eculizumab.27
Sutimlimab, a monoclonal antibody–inhibiting serine protease C1s in the classical complement pathway, recently completed testing in a single-arm phase 3 clinical trial of nontransfused CAD patients (CARDINAL).11,28,29 Patients tolerated the drug well and saw an increase in hemoglobin level of approximately 2.6 g/dL from baseline by week 3, with a hemoglobin level greater than or equal to 11 g/dL maintained from week 3 to the end of the 26-week study period. Notably, the hemoglobin level response in this study was earlier and more robust than studies using eculizumab. Sutimlimab also improved fatigue.11 These findings were validated in a randomized placebo-controlled phase 3 study (CADENZA), with 73% of patients meeting the primary end point of hemoglobin levels rising more than or equal to 1.5 g/dL without the need for transfusion of additional CAD-directed therapies.30 Sutimlimab is being studied in an open-label multicenter trial for transfused CAD patients (NCT03347396). Sutimlimab is the only US Food and Drug Administration-approved drug for adults with CAD to decrease the need for transfusion.
BIVV020, an anti-C1s antibody, is in a phase 1 trial assessing safety and tolerability in CAD patients (clinicaltrials.gov identifier: NCT4269551).29 Pegcetacoplan, a C3 inhibitor, successfully causes the cessation of hemolysis in in vitro models.31 In paroxysmal nocturnal hemoglobinuria clinical trials, it is well tolerated and has superior clinical outcomes compared to eculizumab.32,33 A phase 3, randomized, double-blind, placebo-controlled multicenter trial evaluating pegcetacoplan in CAD is recruiting (NCT05096403).
Unlike rituximab-based protocols, complement-directed treatments require ongoing dosing. Because complement inhibitors do not stop IgM binding and RBC agglutination, ischemic symptoms are not alleviated with this class of drugs, though hemolysis may improve.34 The most appropriate use of complement- directed drugs may be alleviating acute, severe anemia or bridging to durable rituximab-based regimens.
Other promising/experimental therapies
CAD is driven by clonal B-cell expansion that produces IgM. Bruton's tyrosine kinase inhibitors, phosphatidylinositol 3-kinase δ inhibitors, or B-cell lymphoma 2 (BCL2) inhibitors are efficacious in other clonal B-cell disorders and have potential in CAD. Bruton's tyrosine kinase inhibitors such as ibrutinib are effective in both CAD and CAS.35 Daratumumab, an antibody against plasma cell CD38, used in multiple myeloma, has successfully resolved hemolysis, reduced agglutinin titers. and resolved non–complement- mediated symptoms in refractory CAD.36,37
Pediatric cAIHA
cAIHA is rare in children compared to adults. The majority of cases develop secondary to infection, with Mycoplasma pneumoniae being the most common cause. Epstein-Barr virus, influenza, cytomegalovirus, and varicella are also implicated.38 Infectious testing is recommended for all children, and autoimmune screening is suggested (eg, antinuclear antibodies titer) in adolescents and teens. An extensive workup to distinguish CAD from CAS such as that described in Clinical Case 1 is not necessary; cAIHA caused by low-grade clonal B-cell lymphoproliferation is not reported in children. Treatment for pediatric cAIHA is focused on supportive care during the acute presentation while awaiting spontaneous disease resolution. Measures include avoiding cold exposure and warming blood products or other intravenous infusions. Though in this case the child's Mycoplasma pneumoniae was treated, most infections driving pediatric cAIHA are self-limited. In an emergency, plasmapheresis can be utilized. Rituximab and complement-directed therapies have not been studied in pediatric cAIHA.39
Paroxysmal cold hemoglobinuria
Paroxysmal cold hemoglobinuria (PCH) is uncommon in children and exceedingly rare in adults.5 PCH is caused by the Donath-Landsteiner antibody directed against the erythrocyte P antigen.6 IgG fixes complement at cold temperatures. After cooling and rewarming the sample, complement is amplified, and RBCs undergo intravascular hemolysis.5,39 PCH is triggered by viral infections and treatment is supportive. Because it is IgG-mediated, PCH patients may be steroid responsive.39
Conclusion
CAD is a rare cause of AIHA. In adults it is typically primary, driven by clonal lymphoproliferation leading to IgM production, thereby triggering complement. The understanding of CAD pathophysiology is improving, and treatment options have expanded. Novel therapeutics targeting clonal B cells or complement have shown success, and there are more new therapies on the horizon. Increasingly, patients are able to receive therapy to improve anemia, acral symptoms, and fatigue. With more well-tolerated treatment options available, the risk/benefit ratio of treating cAIHA is shifting toward improved symptom control for more patients.
Conflict-of-interest disclosure
Jenny McDade Despotovic: consultancy: Novartis; research funding: Novartis; honoraria: Novartis, Dova, Amgen; royalties: Uptodate.
Taylor Olmsted Kim: consultancy: Novartis.
Off-label drug use
All drugs are off-label for pediatric patients. For adults, sutimlimab is the only FDA-approved therapy for cold agglutinin disease. The rest listed in Table 2 are off-label.

