Abstract
Chronic phase CML (CP-CML) patients who are resistant to 2 or more tyrosine kinase inhibitors (TKIs) have limited therapeutic options and are at significant risk for progression to the blast phase. Ponatinib has been the drug of choice in this setting for the past decade, but when given at full dose (45 mg/d), the risk of serious vascular occlusive events is substantial. Lower doses mitigate this risk but also reduce the efficacy. Emerging data suggest that a high dose of ponatinib is important to achieve response, but a lower dose is usually sufficient to maintain response, introducing a safer therapeutic pathway for many patients. The recent development and approval of the novel allosteric ABL1 inhibitor, asciminib, for CP-CML patients with resistant disease provides another potentially safe and effective option in this setting. These recent therapeutic advances mean that for most resistant CP-CML patients who have failed 2 or more TKIs, 2 excellent options are available for consideration—dose modified ponatinib and asciminib. Patients harboring the T315I mutation are also candidates for either ponatinib or asciminib, but in this setting, higher doses are critical to success. Lacking randomized comparisons of ponatinib and asciminib, the best choice for each clinical circumstance is often difficult to determine. Here we review emerging evidence from recent trials and make some tentative suggestions about which drug is preferable and at what dose in different clinical settings using case studies to illustrate the key issues to consider.
Learning Objectives
Determine appropriate assessments for CP-CML patients being considered for a change in therapeutic approach because of TKI resistance
Recommend the optimal TKI and the dosing regimen for a CP-CML patient with TKI resistance
What disease and patient assessments are indicated for the following 3 patients with resistant chronic phase chronic myeloid leukemia (CP-CML), and what is the best therapeutic approach in each case?
CLINICAL CASE 1
A 32-year-old woman with CP-CML and a high-risk EUTOS Long-Term Survival (ELTS) score received frontline dasatinib at 100 mg/d for 4 months, then switched therapy after confirmed early molecular response failure (>10% BCR::ABL1IS). After 4 months of nilotinib at a dose of 400 mg twice daily, there was no improvement in her molecular response.
CLINICAL CASE 2
A 55-year-old woman with CP-CML and a low-risk ELTS score was started on nilotinib and achieved a major molecular remission (MMR; <0.1% BCR::ABL1IS) by 12 months. After 18 months, she sequentially switched to bosutinib, then dasatinib, both for intolerance issues, but gradually lost MR2 (>1% BCR::ABL1IS).
CLINICAL CASE 3
A 72-year-old man with an intermediate-risk ELTS score failed to achieve MR2 by 12 months on imatinib then received dasatinib at 100 mg/d for 6 months, with an initial transient improvement in response (0.5% BCR::ABL1IS). He then rapidly lost molecular response (15% BCR::ABL1IS).
Introduction
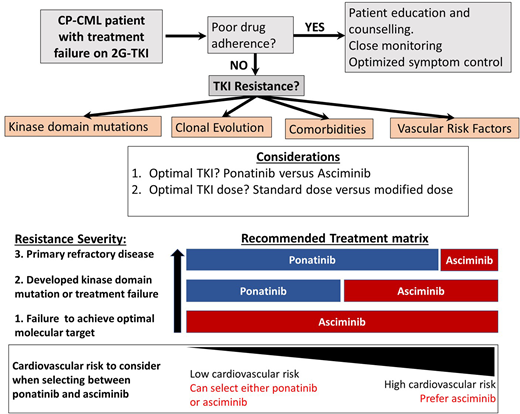
For most patients with CP-CML who have achieved and maintained an optimal molecular response, the ongoing management is fairly straightforward. Ensuring optimal molecular targets are met and maintained, managing drug-related side effects, monitoring drug adherence, and paying careful attention to minimize drug-induced organ toxicities are the main priorities.1,2 At the extremes of response, management can be more complex. Patients achieving and maintaining deep molecular responses over several years are considered candidates for treatment-free remission, although embarking on a treatment-free remission attempt requires extensive discussion and patient education with a careful review of eligibility. Furthermore, once therapy has been ceased, frequent monitoring is essential, as well as a high degree of vigilance looking for molecular relapse.3-6 At the opposite end of the response spectrum, treatment failure to at least 2 tyrosine kinase inhibitors (TKIs) or to just 1 second-generation TKI (2G-TKI) requires expert management while still aiming to maximize optimal responses and also to prevent disease progression and TKI-induced toxicity. Treatment failure to one or more 2G-TKIs infrequently can be salvaged by switching to another 2G-TKI,7-10 usually in cases in which specific ABL1 kinase domain (KD) mutations have emerged. The clinical development of ponatinib has provided an effective option for many of these resistant patients. The PACE study demonstrated its effectiveness in the setting of resistant disease, both in the case of resistance associated with a KD mutation, including the gatekeeper T315I mutation, and in cases without clear identifiable resistance mechanisms.11 The key limitation with ponatinib therapy that emerged in the PACE study and other studies was the very high rate of arterial occlusive events (AOEs) observed, which necessitated the premature termination of some ponatinib-based clinical trials and a transient withdrawal from the market.11,12 In the CP-CML arm of the PACE study, the reported rate of AOEs was 31%.11
The therapeutic challenges related to managing these TKI-resistant CP-CML patients have become more complex recently because we now have safer and arguably better therapeutic options to consider. The recent demonstration from the OPTIC trial of a less toxic but still efficacious dosing regimen for ponatinib has changed the risk/benefit equation, making ponatinib a more attractive option in this setting.13 Equally promising has been the approval of the allosteric ABL1 inhibitor asciminib, which offers good tolerability and high efficacy in similar high-risk settings.14,15 Clinicians now have several options for patients with 2G-TKI resistance, but it is often not clear which is the best drug choice and the optimal dose regimen. The prospects of good disease control and the risks of toxicity with each option need to be carefully assessed. To provide some clarity about how to make the best decision for a particular patient, we explore the emerging findings from recent trials for resistant patients. We also present and discuss several clinical cases to illustrate the value of considering all patient- and drug-specific parameters before selecting the optimal therapeutic approach for each individual patient.
The OPTIC trial
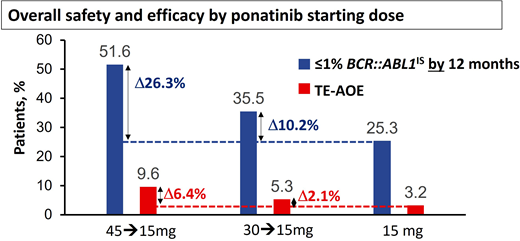
Modeling based on the PACE study and other studies suggested the risk of AOEs was significantly lower at lower doses,11,16 leading to the approach adopted for the OPTIC trial.13 In OPTIC, patients were randomized to receive either 45 mg/d, 30 mg/d, or 15 mg/d as a starting dose.13 In the case of patients in the higher-dosing cohorts, the dose was reduced to 15 mg/d once MR2 (BCR::ABL1IS < 1%) was achieved.13 As predicted, the dosing strategy of 45 mg/d with dose reduction proved to be significantly less toxic in terms of AOEs compared to the PACE data, where doses were often maintained at 45 mg/d throughout.13 Remarkably, this approach using high doses initially and deescalated doses in maintenance also proved to be highly efficacious and significantly superior to the other 2 arms, with an MR2 rate at 12 months of 52% in the 45-mg cohort vs 36% in the 30-mg cohort and 25% in the 15-mg cohort.13 Furthermore, the OPTIC study provides further insights into the delicate balance of efficacy and safety that needs to be considered when selecting the initial dose (Figure 1). In resistant cases with the T315I mutation, the advantage of starting at 45 mg/d over 30 mg/d is significant, with MR2 rates by 12 months at 60% and 25%, respectively.13 There was also an advantage with the 45-mg starting dose compared to the 30-mg cohort in patients with a non-T315I KD mutation (56% vs 40% MR2 by 12 months).13 In patients with resistance or intolerance without a KD mutation, the advantage was less marked (46% vs 38%).12
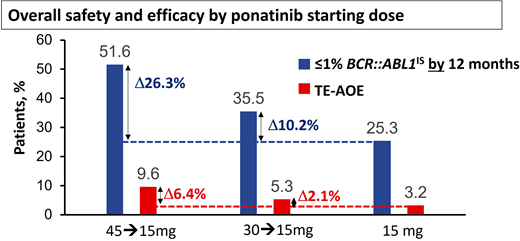
Overall safety and efficacy of ponatinib based on starting dose. The analysis is a descriptive clinical summary of the data to illustrate the relationship between the efficacy and the AOE rate. TE-AOE, treatment-emergent arterial occlusive events. Reproduced with permission from Cortes et al.13
Overall safety and efficacy of ponatinib based on starting dose. The analysis is a descriptive clinical summary of the data to illustrate the relationship between the efficacy and the AOE rate. TE-AOE, treatment-emergent arterial occlusive events. Reproduced with permission from Cortes et al.13
The higher efficacy achieved with higher starting doses needs to be weighed against the higher toxicity. In patients with recent AOEs on a prior TKI, even a brief exposure to full-dose ponatinib may prove very hazardous. Treatment-emergent AOEs per 100 patient-years in OPTIC were 3.2 for the 15-mg cohort, 5.3 for the 30-mg cohort, and 9.6 for the 45-mg cohort.13 To distinguish this dosing strategy from the conventional strategy of maintaining the starting dose if it is well tolerated, we refer to this dosing regimen as ponatinib 45 mg (OPTIC dosing) and ponatinib 30 mg (OPTIC dosing) for clarity.
The ASCEMBL trial
The ASCEMBL trial randomized CP-CML patients who were intolerant or resistant to 2 or more TKIs to either asciminib or bosutinib in a 2:1 ratio.15 Asciminib dosing was set at 40 mg twice daily based on the tolerability assessments of a number of dosing levels in the phase 1 study and pharmacokinetic modeling demonstrating that either 40 mg twice daily or 80 mg/d of asciminib exceeded the 90% threshold for mean inhibitory concentrations.14 It demonstrated a clearly superior rate of MMR (BCR::ABL1IS < 0.1%) by 6 months for asciminib (26% vs 13%).15 However, MR2 may be a more realistic target in this multiresistant setting. The difference was significant here as well—by 12 months, 42% vs 19% had achieved MR2.9 Another major difference between the 2 arms was the rate of discontinuation. In the most recent update,17 43% of patients on the asciminib arm discontinued therapy, including 24% for lack of efficacy and 6% for adverse events (median follow-up, 19 months). In contrast, 78% were no longer taking bosutinib, with 36% stopping due to lack of efficacy and 24% because of an adverse event.17 There were 7 patients on the asciminib arm who had AOEs at the most recent analysis, which represents a rate of 3.4 per 100 patient-years.17 All 7 patients had received nilotinib previously, and 3 had received ponatinib.17 Nonetheless, the observed rate of AOEs was higher with asciminib compared with bosutinib. ASCEMBL demonstrated the clear superiority of asciminib over bosutinib in the resistant and intolerant third-line setting.
Comparing ponatinib and asciminib trial data
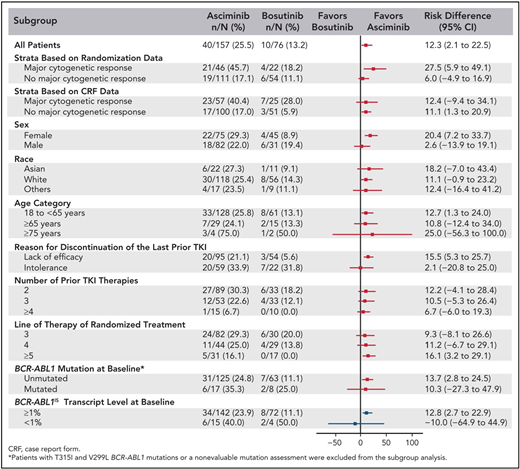
The best way to determine whether ponatinib or asciminib is superior as a third-line option in different clinical settings would be to conduct head-to-head randomized studies. However, as these have not been initiated at this stage, we present our opinions based on the data available from the phase 1 asciminib trial, the ASCEMBL trial, and the OPTIC trial to formulate an initial assessment regarding which drug may be preferential in specific cases (Table 1). OPTIC recruited a higher proportion of patients with KD mutations and a higher proportion of patients who failed to achieve any molecular response to their prior TKI.13 In comparison, ASCEMBL had a higher proportion of intolerant patients compared to OPTIC. Overall response rates look similar when comparing the 45-mg cohort of OPTIC to the asciminib arm of ASCEMBL.13,17 In patients without T315I, the 12-month rates of MR2 were similar; 42% for asciminib and 49% for the ponatinib 45-mg cohort.13,15 However, response rates appear to be more variable according to the baseline response in the asciminib cohort. Notably, only 17% of the patients not in a major cytogenetic response (MCyR) at baseline achieved MMR at 6 months, compared to 40% of patients who were in MCyR at baseline (Figure 2).15 Moreover, the current accepted standard dose of asciminib of 40 mg twice daily may be inadequate to combat highly resistant CML, as evidenced by the higher doses required in T315I-mutated patients.14 Comparable data from OPTIC are not available, but in the 45-mg ponatinib cohort, the MR2 rates were 50% by 12 months regardless of prior response on historical TKI therapy.13 On this (tentative) basis, ponatinib may be a better choice in highly resistant cases, and asciminib, with its superior safety profile, may be preferable in patients with less profound resistance.
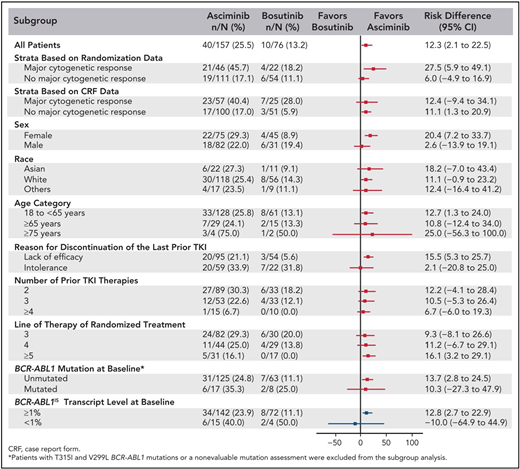
Subgroup analysis of the ASCEMBL study: risk difference with 95% CIs for MR3 achievement by 24 weeks of either asciminib or bosutinib. CRF, case report form. Reproduced with permission from Rea et al.15
Subgroup analysis of the ASCEMBL study: risk difference with 95% CIs for MR3 achievement by 24 weeks of either asciminib or bosutinib. CRF, case report form. Reproduced with permission from Rea et al.15
Another important comparison is the risk of AOEs with each agent. Many variables make this comparison problematic. These include different eligibility criteria regarding cardiac risk factors and previous cardiovascular history, different duration of follow-up, and different criteria for determining a treatment-emergent AOE. Acknowledging these limitations seems to show an advantage for asciminib, but this needs careful assessment with ongoing data from these and other studies. Rates of AOEs per 100 patient-years were 9.6 for the ponatinib 45-mg cohort in OPTIC compared to 3.4 for asciminib in the ASCEMBL trial.
Optimal therapy for patients with T315I mutation
Both ponatinib and asciminib are active against the T315I mutation, but in both cases higher doses are important for efficacy. In the OPTIC trial, 15 of 25 (60%) patients with T315I receiving the 45-mg initial dose achieved MR2 by 12 months, compared to 5 of 20 (25%) who started at 30 mg/d.13 Asciminib efficacy against the T315I mutant was assessed in the phase 1 trial, but response rates were low until doses above 150 mg twice daily were given.14 For that reason an expanded cohort of 48 CP-CML and 4 acute-phase CML patients with the T315I mutation were given asciminib at 200 mg twice daily.18 At this dose, molecular responses were frequent, and toxicity appeared similar to standard -ose asciminib (Table 2). With a median follow-up of 17 months, 35 of 52 (67%) remained on therapy, and 23 of 49 (47%) who were not in MMR at baseline achieved MMR by 24 months.18 This is impressive, especially considering that 28 of 52 (54%) had prior therapy with ponatinib and had developed either intolerance or resistance. In ponatinib-naive patients, the rate of MMR was 57% by 6 months, compared to 29% in ponatinib-resistant/intolerant patients.18
Thus, both drugs are highly active when used at the correct dose for patients with the T315I mutation. When patients on ponatinib 45 mg/d deescalated dosing as per the OPTIC protocol, the overall rate of loss of response exceeded 30% (the actual rate was not specified in the OPTIC paper).13 Therefore, a risk assessment should be undertaken before dose deescalating in the setting of ponatinib therapy for patients with the T315I mutation. In support of asciminib as the first choice in the T315I setting, tolerance appears superior to ponatinib 45 mg (OPTIC dosing), while efficacy appears similar (Table 2).18 In support of ponatinib as the first choice, we have more data and longer follow-up to reassure us that responses are stable and durable in T315I patients. Since there is no clear “winner,” the choice should be individualized by weighing the disease-related factors and the cardiovascular risk.
Optimal therapy in the case of other KD mutations
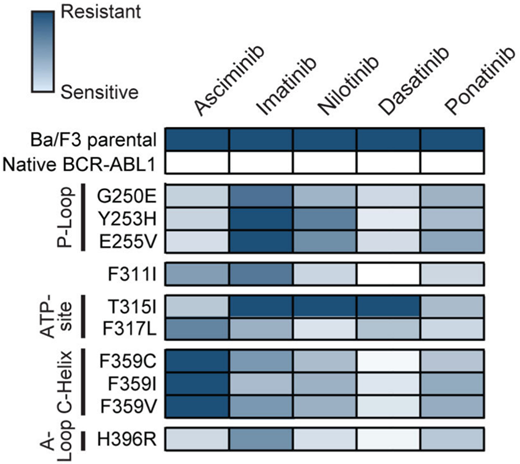
Both ponatinib and asciminib are active against most adenosine triphosphate-site KD mutations,19,20 although sensitivity is generally lower than it is for unmutated BCR::ABL1. KD mutations of concern for asciminib include F359V/C/I, which either emerged or persisted without evidence of molecular response in several cases in ASCEMBL and the phase 1 study.15,21 This was predicted to be a partially resistant mutation in in vitro studies.21 More data are needed to determine asciminib activity against the F317L mutation, which appears to induce a degree of asciminib resistance based on in vitro studies (Figure 3).21 However, in all presented scenarios, higher doses of asciminib may be required for optimal efficacy, analogous to the higher doses necessary to combat the T315I mutation.
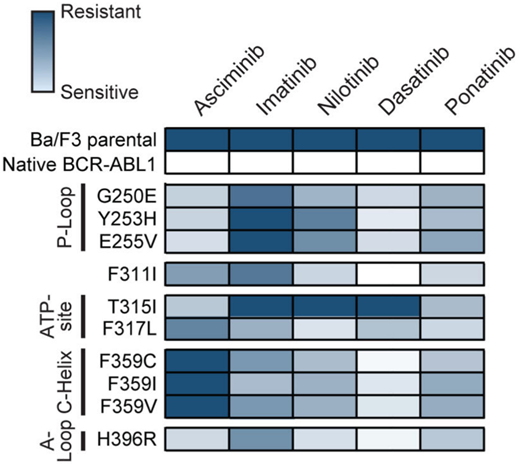
Heat map summary of IC50 values for TKIs against a panel of Ba/F3 cell lines expressing KD mutations. ATP, adenosine triphosphate. Reproduced with permission from Eide et al.21
Heat map summary of IC50 values for TKIs against a panel of Ba/F3 cell lines expressing KD mutations. ATP, adenosine triphosphate. Reproduced with permission from Eide et al.21
Any role for 2G-TKIs in this setting?
In most cases the optimal choice of next-line therapy is between ponatinib and asciminib. There are specific cases in which a 2G-TKI may still have a role. This would include cases in which resistance is driven by a mutation that is well covered by another 2G-TKI. For instance, the development of V299L or F317L mutations is highly sensitive to nilotinib and bosutinib, and F359V/I/C mutations are highly sensitive to both dasatinib and bosutinib (Figure 3).2
Additionally, in some countries eligibility rules prevent access to sequential therapy with either asciminib or ponatinib in the setting of 2G-TKI failure without developing resistance or intolerance to an alternative 2G-TKI. In this setting, a 3-to-6 month trial is generally sufficient to determine if the newly commenced 2G-TKI is likely to be effective and salvage the situation. It is rare for patients who have no improvement in molecular response after 6 months of a salvage therapy to eventually achieve MR2 or better with longer-term exposure.
The intolerant patient
The focus of this chapter is TKI resistance, but it is worth commenting on the challenging patient who is intolerant to 2 or more TKIs. In this setting the efficacy and tolerability of asciminib look very favorable based on the phase 1 asciminib study,14 and in most cases, ponatinib would not be the primary drug of choice due to its higher rates of intolerance and toxicity. Whether lower doses of ponatinib could achieve comparable response and tolerance to asciminib in these patients, where the predominant challenge is intolerance, remains to be established.
Future options
Targeting 2 distinct regions on ABL1, the adenosine triphosphate- and myristate-binding sites, with asciminib and conventional TKIs may also be a future option to combat resistance, with the phase 1 clinical trial yet to formally report outcomes for the relevant treatment arms that investigated combination therapy.20 Furthermore, murine models have demonstrated that combining asciminib with ponatinib can inhibit the growth of T315I-containing compound mutations.21 A number of novel BCR::ABL1–targeting agents are under investigation for resistant CML. One such drug is vodobatinib, a novel 3G-TKI with limited off-target activity.22 The phase 1 dose escalation study recently reported promising safety and efficacy results generated in both ponatinib-naive and ponatinib-treated patients.22 Furthermore, cardiovascular toxicity did not appear to be a prominent feature at this early stage, which may distinguish vodobatinib from the other available agents for resistant CML. Similarly, another 3G-TKI, olverembatinib, has also had early phase 1 results reported in first- and second-generation-resistant CP-CML and acute-phase CML.23 Efficacy was observed regardless of the study entry response, predominantly in T315I- mutated patients.23 Responses were less impressive in the absence of T315I. While longer-term follow-up in larger data sets is required, these agents may offer future options for this resistant patient cohort.
CLINICAL CASE 1 (Continued)
This 32-year-old woman with CP-CML and a high-risk ELTS score received frontline dasatinib at 100 mg/d for 4 months, then switched to nilotinib at 400 mg twice daily for 4 months after confirmed early molecular response failure. After a total of 8 months, her BCR::ABL1IS remained over 10%, having never fallen below 10%.
Cytogenetics at 8 months did not show evidence of clonal evolution, but 18 of 20 metaphases were Ph+. No KD mutation was identified at 4 or 8 months. She had no cardiovascular risk factors or significant comorbidities.
Options
Continue nilotinib and review at 12 months.
Switch to bosutinib.
Switch to ponatinib 45 mg/d (OPTIC dosing).
Switch to asciminib.
Proceed to an allograft.
Discussion of options
1. Continue nilotinib: Unless there are significant interruptions to therapy due to toxicity issues or poor adherence that resolve or are overcome, there is very little prospect that the molecular response will improve by continuing the current therapy. In this setting, the risk of disease progression is high. One important consideration is the need to identify those patients who will not achieve response with any TKI as soon as possible so that an allograft can still be scheduled in the chronic phase, where there is still a good prospect of cure.
2. Switch to bosutinib: The likelihood of a response if a third 2G-TKI is given is very low. In ASCEMBL, only 19% achieved MR2 by 12 months on the bosutinib arm, and the response rate was even poorer in patients who were not in an MCyR at baseline, as in this case.
3. Switch to ponatinib 45 mg/d (OPTIC dosing): Given the complete lack of response after 8 months of potent TKI therapy, this may be the best option. In OPTIC, 50% of patients who were not in MCyR at baseline who received 45 mg/d of ponatinib achieved MR2 by 12 months. If a patient is given ponatinib and does not achieve MR2 after a further 6 months, the prospect of achieving this target with ongoing ponatinib is low, and hence switching to asciminib or proceeding to an allograft would be appropriate.
4. Switch to asciminib: As discussed earlier, responses to asciminib are probably inferior to ponatinib 45 mg/d (OPTIC dosing) in patients with little or no response to prior therapy. The exception would be in cases in which the risk of a vascular event was high, where asciminib (or lower-dose ponatinib) may be preferred.
5. Proceed to an allograft: While an allograft is not directly recommended at this stage, it would be prudent to tissue type the patient and identify an allogeneic donor in case of failure to respond to third-line therapy. In the event of persistent therapy failure, the patient can proceed to an allogeneic stem cell transplant promptly without delay.
Best option: ponatinib 45 mg daily with a dose reduction to 15 mg daily if MR2 is achieved.
CLINICAL CASE 2 (Continued)
The 55-year-old woman with CP-CML and a low-risk ELTS score was started on nilotinib and responded well, achieving MMR by 12 months. After 18 months, she developed peripheral vascular disease and switched to bosutinib and then dasatinib due to further toxicity but sequentially lost MMR and MR2 after 3 years.
Further assessments: There were no KD mutations or evidence of clonal evolution on marrow examination. There were substantial interruptions secondary to adverse events as well as periods of poor drug adherence. She had peripheral vascular disease and well-controlled hypertension.
Options
Switch to imatinib.
Switch to ponatinib 45 mg/d with dose reduction once MR2 is achieved.
Switch to ponatinib 30 mg/d with dose reduction once MR2 is achieved.
Switch to asciminib 40 mg twice daily.
Discussion of options
1. Switch to imatinib: It is possible that her loss of response is more related to extensive interruptions to therapy and poor adherence rather than drug resistance, so imatinib may still be an effective drug in this setting. However, given her intolerance to all 3 2G-TKIs, imatinib may not be well tolerated, and her adherence may be poor once again.
2. Switch to ponatinib 45 mg/d (OPTIC dosing) with dose reduction once MR2 is achieved: While she has lost MR2, it is still very possible that she still has TKI-sensitive disease, so she would likely have an excellent response to full-dose ponatinib. However, the risk of AOEs is still significant at this dose, especially given her history, and other options may be equally or more efficacious without the same level of risk.
3. Switch to ponatinib 30 mg/d (OPTIC dosing) with dose reduction once MR2 is achieved: This dose provides a lower risk of AOEs (5.3 events per 100 patient-years compared to 9.6 for the 45-mg ponatinib cohort in OPTIC). However, the rate of molecular response is lower than with the higher dose of ponatinib. In patients with no KD mutations, 46% and 38% achieved MR2 by 12 months in the ponatinib 45-mg and 30-mg dose cohort, respectively. This may not be a major difference as progression-free and overall survival were not substantially inferior with the modified dose. This would be an acceptable option except for the fact that the patient has a history of peripheral vascular disease.
4. Switch to asciminib at 40 mg twice daily: This would be a relatively safe and effective option in this setting. Patients in MCyR in ASCEMBL who received asciminib had a 40% rate of MMR by 6 months. The risk of AOEs (per 100 patient-years) in this cohort was 3.4.17 Given her consistent tolerability issues with all 3 2G-TKIs, asciminib would be the option most likely to be well tolerated.
Best option: Asciminib at 40 mg twice daily and consider a switch to ponatinib if MR2 is not achieved after 6 to 12 months. Imatinib may be another option, but given the loss of responses on previous therapy, asciminib would be preferentially recommended.
CLINICAL CASE 3 (Continued)
A 72-year-old man with intermediate-risk ELTS failed to achieve MR2 by 12 months on imatinib. He then received dasatinib at 100 mg/d for 6 months with an initial improvement in response (best response, 0.5% BCR::ABL1IS) but then lost molecular MR2 (15% BCR::ABL1IS).
Further assessments: He had no mutation detected when switching to dasatinib, but when he lost MR2, there was evidence of emergence of the F317L mutation. From a cardiovascular risk perspective, he had a single coronary stent inserted 10 years ago and has poorly controlled hypertension.
Options
- 5.
Switch to bosutinib.
- 6.
Switch to nilotinib.
- 7.
Switch to ponatinib 45 mg/d (OPTIC dosing).
- 8.
Switch to ponatinib 30 mg/d (OPTIC dosing).
- 9.
Switch to asciminib 40 mg twice daily.
Discussion of options
1. Switch to bosutinib: The F317L mutation would appear to be sensitive to bosutinib, but in this patient there was also evidence of resistance to imatinib that was not driven by a KD mutation. Bosutinib may still be a reasonable choice in this setting, especially given its lower risk of AOEs. However, the experience in ASCEMBL would suggest that it may be poorly tolerated.
2. Switch to nilotinib: The F317L mutation is sensitive to nilotinib, but the risk of vascular events is probably too high, especially since a higher dose is likely to be needed (400 mg twice daily).
3. Switch to ponatinib 45 mg/d (OPTIC dosing): Patients in the OPTIC study with a non-T315I mutation who received the higher initial dose of ponatinib had a rate of MR2 by 12 months of 56%. The main concern would be the AOE risk given the patient's history and poorly controlled hypertension. This patient would not have been eligible for the OPTIC trial, so the rate of AOEs would likely be significantly higher than the rate observed on this arm of the OPTIC study.
4. Switch to ponatinib 30 mg/d (OPTIC dosing): Given the concern about the risk of AOEs in this patient, even this modified dose represents a significant risk. The rate of AOEs per 100 patient-years was 5.3 for this dose in OPTIC, but this patient would not have been eligible for OPTIC, so the AOE risk is probably higher.
5. Switch to asciminib 40 mg twice daily: There are very limited data regarding the sensitivity of the F317L mutation to asciminib. In vitro studies suggest a level of resistance greater than that seen with the T315I mutation. This mutation was only found in 2 patients at baseline in the asciminib arm of ASCEMBL; neither had a molecular response.15 Along with the F359C/I/V mutation, we need more clinical data before we can determine whether asciminib is effective in this setting and, if so, at what dose.
Best option: bosutinib may be the safest option, with a switch to ponatinib at a modified dose if no molecular response occurs after 3 to 6 months.
Acknowledgments
Timothy P. Hughes received support from the National Health and Medical Research Council of Australia (APP1135949) and has the financial support of Cancer Council SA's Beat Cancer Project on behalf of its donors and the state government of South Australia through the Department of Health.
Conflict-of-interest disclosure
Timothy P. Hughes: research funding: Novartis, Bristol Myers Squibb; honoraria: Takeda, Novartis, Bristol Myers Squibb.
Naranie Shanmuganathan: research funding: Novartis; honoraria: Takeda.
Off-label drug use
Timothy P. Hughes: nothing to disclose.
Naranie Shanmuganathan: nothing to disclose.