Abstract
Diamond-Blackfan anemia (DBA) is an inherited bone marrow failure syndrome, characterized as a rare congenital bone marrow erythroid hypoplasia (OMIM#105650). Erythroid defect in DBA results in erythroblastopenia in bone marrow as a consequence of maturation blockade between the burst forming unit–erythroid and colony forming unit–erythroid developmental stages, leading to moderate to severe usually macrocytic aregenerative (<20 × 109/L of reticulocytes) anemia. Congenital malformations localized mostly in the cephalic area and in the extremities (thumbs), as well as short stature and cardiac and urogenital tract abnormalities, are a feature of 50% of the DBA-affected patients. A significant increased risk for malignancy has been reported. DBA is due to a defect in the ribosomal RNA (rRNA) maturation as a consequence of a heterozygous mutation in 1 of the 20 ribosomal protein genes. Besides classical DBA, some DBA-like diseases have been identified. The relation between the defect in rRNA maturation and the erythroid defect in DBA has yet to be fully defined. However, recent studies have identified a role for GATA1 either due to a specific defect in its translation or due to its defective regulation by its chaperone HSP70. In addition, excess free heme-induced reactive oxygen species and apoptosis have been implicated in the DBA erythroid phenotype. Current treatment options are either regular transfusions with appropriate iron chelation or treatment with corticosteroids starting at 1 year of age. The only curative treatment for the anemia of DBA to date is bone marrow transplantation. Use of gene therapy as a therapeutic strategy is currently being explored.
Learning Objectives
Recognize a case of DBA
Manage anemia and potential clinical complications of DBA
CLINICAL CASE
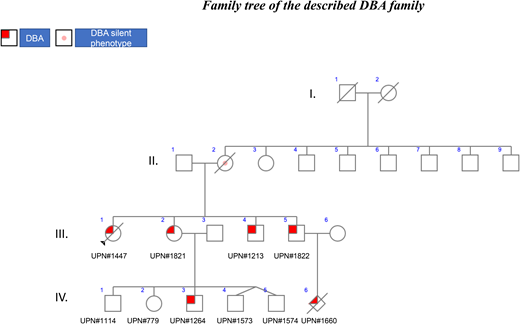
The family described below is an excellent illustration of the various issues that are encountered in the diagnosis and treatment of Diamond-Blackfan anemia (DBA) due to incomplete penetrance.
The family is the largest of the 417 DBA families registered in the French DBA registry (Observatoire Français de l’Anémie de Blackfan-Diamond) and includes 3 generations of affected patients (Figure 1). In this family, the mode of inheritance is familial, as is the case for 45% of the DBA-affected patients registered in the different registries around the world. In 55% of individuals, DBA is the result of sporadic or de novo inheritance. Interestingly, in the illustrated family, all 4 children, 2 girls and 2 boys, developed DBA with different phenotypes (Figure 1). Of note, in the 1980s, no DBA gene had been identified, and steroids were started following DBA diagnosis in all instances, which is not the recommended treatment currently: steroids should be initiated only after the first year of life.1
The child, UPN#1447, at age 2.3 years with normal white and platelet cell counts, was initially diagnosed with transient erythroblastopenia of childhood (TEC) on the basis of an isolated severe normochromic, macrocytic mean corpuscular volume (MCV = 91.4 fL) aregenerative (7.5 × 109/L reticulocytes) anemia (hemoglobin (Hb) level at 79 g/L) with erythroblastopenia documented on the bone marrow smear (1% of total erythroblasts [Eb, normal 5%-30%]). Bone marrow cellularity was normal with no signs of dysplasia. Parvovirus B19 serology was negative for both immunoglobulin M and immunoglobulin G. The patient recovered with no treatment except for a short course of steroid treatment (for a few weeks) with no red cell blood transfusions. At the same time, increased expression of hemoglobin F (HbF) at 9% (normal <2% at 6-24 months of age) along with a significantly elevated erythrocyte adenosine deaminase (eADA) activity at 5.95 nmol/min/mg Hb (or U/g Hb) (normal: 1.50 ± 0.2) has been reported. UPN#1447, initially diagnosed with TEC, has been finally diagnosed with DBA after the identification of a RPS19 gene mutation. In retrospect, the association of pure red blood cell hypoplastic anemia, in conjunction with an increased percentage of HbF and eADA activity, and response to steroid was in accordance with this diagnosis.1,2 However, it is to be noted some clinicians consider TEC to be one of the DBA phenotypes with a low penetrance.3 TEC and DBA should be differentiated, but it is sometimes very difficult to discriminate between the 2 phenotypes as only the time course of the evolution of anemia can clearly distinguish between them (see Table 1 for differential diagnosis between TEC and DBA). In order to be cautious, we recommend a molecular screening and the careful follow-up of any child with TEC or inform the parents to bring back the child for further evaluation in case of recurrence of the anemia (Table 2).
UPN#1821 was the second child of the family, a girl born 2 years later. In contrast to UPN#1447, UPN#1821 exhibited a 2-month-old, severe anemia with a nadir of 20 g/L Hb. The anemia was normochromic and macrocytic (high MCV of 114.8 fL), which is classical for DBA, and with a modest degree of regeneration (60 × 109/L reticulocytes). The characteristic erythroblastopenia was noted on the bone marrow smear with only 1% of Eb. UPN#1821 exhibited features of fetal erythropoiesis with HbF at 18% and a large increase in eADA activity at 5 nmol/min/mg Hb. A valgus foot deformity was noted as the only congenital malformation. UPN#1821 received a regular dose of 2 mg/kg/d of steroid for a month, which allowed for transfusion independency, and was subsequently maintained on low-dose steroid therapy (0.1 mg/kg/d). However, she became later steroid resistant, and she is currently being regularly transfused every 3 weeks (Table 2).
UPN#1822 was diagnosed with DBA at 1 month of age. Surprisingly at this time, the bone marrow smear did not exhibit the characteristic DBA erythroblastopenia with 31% of erythroid precursors. However, a large increase in eADA activity of 4 nmol/min/mg Hb was noted. Posterior hypospadias was noticed. He received an initial dose of steroid at 2 mg/kg/d, and the dose was gradually decreased to 0.2 mg/kg/d, which enabled the stabilization of the Hb level. The steroid treatment lasted for 18 years and 10 months, at which point the patient became steroid independent and maintains a reasonable Hb level with no need for steroids (Table 2).
Finally, UPN#1213, the fourth child of this family, was also a DBA-affected patient. He was diagnosed at 1 month and 3 weeks of age with erythroblastopenia with 4% of Eb in the bone marrow. Like his siblings, his eADA activity was increased to 4.38 nmol/min/mg Hb. Hypospadias was once again noticed. He was treated with an initial dose of 2 mg/kg/d of steroid, which normalized the Hb level. He is now 32 years old and still undergoing steroid therapy at 10 mg per day. At the last follow-up, he exhibited a moderate macrocytic (MCV at 105.9 fL) anemia with an Hb level at 108 g/L with 38.6 × 109/L reticulocytes, in conjunction with normal white blood cell and platelet counts (Table 2).
Clinical and biological presentation of DBA: take-home message
DBA is usually characterized by a moderate to severe, macrocytic aregenerative anemia. The other cell lineages are usually normal, but on occasion, neutropenia, thrombocytopenia, and, in some instances, even thrombocytosis may be noted at the time of diagnosis. The erythroblastopenia (<5% of erythroid precursors) in an otherwise normal bone marrow (no dysplasia and normal cellularity) on the bone marrow smear or pure hypoplastic anemia on bone marrow biopsy can confirm the diagnosis. Bone marrow examination is mandatory to avoid misdiagnosis. Erythroblastopenia is the consequence of blockade in erythroid differentiation between the burst forming unit–erythroid and colony forming unit–erythroid progenitor stages.4 The other biological features include increased eADA activity, which is elevated in 90% of the nontransfused patients with DBA and the persistence of features of fetal erythropoiesis (high HbF percentage). They should both be measured at diagnosis before transfusion or at least 3 months following transfusion (Table 3). In 50% of the patients with DBA, various malformations are reported mostly in the cephalic area and the extremities, with the classical but rare triphalangeal thumbs (Table 4).1,5
Going back to the herein reported cases, the molecular screening of the proband (UPN#1447) and her sister (UPN#1821) and 2 brothers (UPN#1822 and UPN#1213) identified a heterozygous 4–base pair deletion near the donor splice site of exon 2 in the RPS19 gene (NM_001022.3: c.71 + 3_71 + 6del; p.?). This variant has indeed been found in the putative TEC case (UPN#1447), and it should have been enough to rule out TEC. This allelic variation was found in the nonanemic (Hb 121 g/L; 78.4 × 109/L reticulocytes) mother, who is considered a so-called silent phenotype, another distinct feature of DBA. In addition, the mother exhibited a normal MCV (96 fL) and an elevated eADA level (4.21 nmol/min/mg Hb). Silent DBA phenotype individuals could be either the parent or a sibling of a DBA proband: they are not anemic but may exhibit a macrocytosis, an elevated eADA, and/or a mutation in a ribosomal protein (RP) gene. Patients with the “silent phenotype” have a risk of DBA complications such as malignancies and may transmit the disease as demonstrated in this family: the mother has transmitted the variant to all 4 of her offspring, reinforcing the dominant inheritance in this family. DBA has been diagnosed in 2 members of the third generation in this family. UPN#1822 experienced a fetal loss (UPN#1660) in utero at 23 weeks +3 days of a female nondysmorphic fetus exhibiting a severe intrauterine growth retardation (weight: 436.5 g, which is between the 5th and the 10th percentiles; head circumference: 18 cm, 5th percentile; height [vertex/coccyx]: 20 cm, 50th percentile for 23--week amenorrhea) with oligohydramnios but without hydrops fetalis. Interestingly, the placenta exhibited defects in the vessels with stenosis, and some villi were hypovascularized. However, no erythroblastopenia was noted. The same allelic variation has been identified in this fetus. We assume that the major growth retardation in relation with DBA may be the origin of the fetal death in utero (Table 5). Hydrops fetalis is a feature of DBA, and its frequency is likely underestimated. Since our description of the first case of sporadic fetal loss due a mutation in the RPS19 gene,6 we subsequently identified additional fetal cases mostly in association with the RPS19 gene and also in association with the RPL15 gene.7 These findings reinforce, as we stated earlier,8 that major complications can occur during pregnancy and the importance of the follow-up of the mother and her offspring during pregnancy with putative aspirin treatment in order to prevent the placenta vascular complications as noted in our case.8
In addition to UPN#1822, UPN#1821 also had 5 offspring, among them twins (UPN#779, UNP#1114, UPN#1264, UPN#1573, UNP#1574) (Figure 1). Prenatal diagnosis was declined, and out of the 5 children, only 1 boy was affected by DBA and carried the same familial allelic variation in the RPS19 gene. UNP#1264 was born at a normal gestational time of 40 weeks with normal mensuration but a conjunctival and skin pallor, tachycardia, and systolic heart murmur due to anemia at 120 g/L (normal value >140 g/L), which decreased to 75 g/L at day 18 after birth. He was transfused with 70 mL of red blood cells. He has since been regularly transfused every 3 to 4 weeks. He did not present any congenital abnormalities. After 1 year as recommended, steroid therapy was started, but there was no response. Chelation therapy with deferasirox was started but had to be interrupted due to liver toxicity. Deferoxamine treatment (1000 mg/d) by nightly infusion over 10 hours, 5 out of 7 days, was started to manage iron overload. Stem cell transplantation was contemplated, and the siblings were tested for the known familial allelic variation, which was not found in any of them. Unfortunately, none of them was HLA identical. He is currently 6 years old and has been recently undergone transplantation with a fully matched unrelated donor.
DBA treatment: take-home message
DBA treatment in 2021 still relies on either chronic blood transfusions (Hb concentration to be maintained >90 g/L) with optimal iron chelation therapy started when the ferritin level is >500 µg/L or treatment with corticosteroids.9-11 The corticosteroids (prednisone or prednisolone) are to be introduced only after the first year of life in order to enable the maximal growth because short stature is part of the malformative syndrome. The corticosteroids are introduced at a dose of 2 mg/kg/d, and their efficacy is usually seen starting at 2 weeks following initiation of therapy, reflected by increased reticulocyte count and Hb level. If there is no response to steroid therapy after 1 month, there is no reason to continue this therapy. In case of efficacy, the corticosteroid should be gradually decreased to a minimum dose that can maintain the Hb level above 90 g/L. The maximal continuous corticosteroid dose must be <0.3 mg/kg/d (<0.5 mg/kg/d in the countries where access to transfusions is difficult or dangerous). In case of corticoresistance or corticodependence (>0.3 [or >0.5 mg/kg/d]), transfusions with iron chelation are currently the only alternative option of treatment,9-11 and hematopoietic stem cell transplantation (HSCT) is indicated following the availability of either an HLA-identical sibling in whom DBA or the silent phenotype has been excluded12-15 or with a fully matched (10/10) unrelated donor.14,16 HSCT should be performed ideally before the age of 5 years and certainly <10 years to avoid HSCT complications. HSCT is also indicated in patients with clonal evolution.12-15 An alternative source of hematopoietic stem cells should be considered as experimental approaches but may be indicated for patients with clonal evolution. HSCT cures anemia and prevents the risk of myelodysplastic syndrome (MDS) and leukemia, but patients with DBA still need to be carefully monitored for the risk of posttransplant solid tumors.17
Last, leucine has been found to be effective in small number of patients with DBA.18 New therapeutic options such as gene therapy are probably the most promising option,19,20 along with other targeted therapies.21 Recently, some innovative therapies have been proposed such as calmodulin inhibitors16 and metformin,22 and it is likely that other new therapeutic options will emerge from translational research (trial NCT03966053 with trifluoperazine).23-26
Genetics in DBA: take-home message
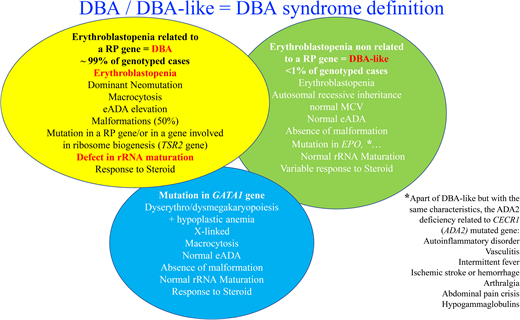
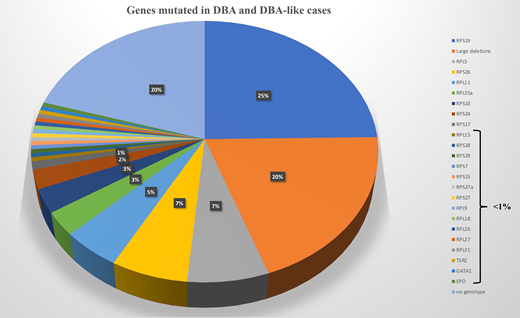
An allelic variation, always in a heterozygous state in a RP, is found in approximately 70% to 80% of the DBA-affected cases.27-30 Mutations in 20 RP genes have been identified (Figure 2). The most frequently mutated gene and the one identified in our clinical case is the RPS19 gene, which accounts for 25% of DBA cases globally.31 Large deletions in these RP genes have been found in approximately 20% of the DBA cases, mostly in RPS17, RPL35a, and RPS19 genes.32 Thus, at least 70% of the patients with DBA carry an allelic variation, including a large deletion in only 8 RP genes, namely, RPS19, RPL5, RPS26, RPL11, RPL35a, RPS10, RPS24, and RPS17 (Figure 2). Multiple pathogenic RP mutations in a patient with DBA have not been reported to date. Phenotypic/genotypic correlation is not readily evident, although it is well established that patients with DBA carrying a RPS19 gene mutation exhibit less malformation compared with others but appear to exhibit a more severe hematologic phenotype and are frequently managed by a transfusion program.33 Neutropenia is more frequently associated with RPL35a,34 cardiac anomalies with RPS24,35 cleft palate with RPL5, and thumbs anomalies with RPL1136 gene mutations. Other RP genes have been found to be mutated in small subsets of DBA-affected patients, each affecting <1% of DBA cohorts (Figure 2). A mutation in a RP gene involved in DBA is associated with defective ribosomal RNA (rRNA) maturation, which is considered the signature feature of the DBA disease, and its documentation confirms the pathogenicity of allelic variation of unknown significance. DBA-like disease is responsible for anemia with erythroblastopenia and a certain degree of steroid response but with the absence of a defect in rRNA maturation. These DBA-like diseases are related to EPO37 and GATA-138,39 gene mutations. The DBA and DBA-like diseases constitute the DBA syndrome. Despite erythroblastopenia and a certain degree of steroid response, ADA2 deficiency related to CECR1 or ADA2 gene mutations is not considered to belong to DBA syndrome (Figure 2).30,40,41
The DBA family we described has been affected by malignancies. The mother, who represents a DBA silent phenotype, was diagnosed with breast cancer that was rapidly fatal. The oldest offspring, UNP#1447, carrying the familial RPS19 allelic variation but never needing any treatment, was diagnosed at age 36 years with a rectal adenocarcinoma and passed away following 16 months of treatment. These observations point to the fact that identifying patients with DBA (and not misdiagnosing them with TEC) is important and that the so-called silent DBA phenotypes should be considered in patients with DBA in their follow-up, particularly for the risk of malignancies. A recent study established a risk of 5% for malignancies in nontransplanted patients with DBA in the National Cancer Institute cohort17 and in the American DBA registry42,43 with an observed/expected ratio of 4.8 for any malignancy, 44.7 for colon carcinoma, 9.4 for lung cancer, 42.4 for osteogenic sarcoma, 352 for MDS, and 28.8 for acute myeloid leukemia.42,43 However, compared with the other inherited bone marrow failure syndromes, the observed/expected ratio is lower for DBA.17 To date, there are no guidelines for MDS/acute myeloid leukemia and solid tumor screening, except recent preliminary recommendations on colorectal carcinoma,44 but this may be warranted and is currently being discussed among DBA cooperative groups.
In conclusion, DBA is a fascinating and complex erythroid disorder, as illustrated from the study of the DBA family we described. Progress is being made in our understanding of the molecular basis for DBA, pathophysiology of the disease, and developing and pursuing new therapeutic options. We anticipate that these advances will enable better clinical management of the patients with DBA in the coming years.
Acknowledgments
Our special thanks go to the affected individuals, their families, and the DBA French patient association (Association Française de la Maladie de Blackfan-Diamond [AFMBD], president Marcel Hibert). We sincerely thank our 2 mentors, Gil Tchernia and Mohandas Narla, for their support, friendship, and many fruitful scientific discussions. We also thank all our collaborators in France and around the world involved in the mutational screening analysis in France (Cécile Masson, Christine Bole, and the team from the Genomics Core Facility, Institut Imagine-Structure Fédérative de Recherche Necker, and Anaëlle Jaouen, Ludivine David, Julie Galimand, and Hélène Bourdeau, technicians from the hematology lab in R. Debré hospital, Paris). L.M.D.C. is supported by #ANR EJPRD/ANR-19-RAR4-0016 (European Union's Horizon 2020 research and innovation program under the EJP RD COFUND-EJP No 825575), the Laboratory of Excellence for Red Cells ((LABEX GR-Ex)-ANR Avenir-11-LABX-0005-02), and the French National PHRC OFABD (DBA registry). T.M.L. and L.M.D.C. are supported by the ANR grant EJPRD DBAGenCure (coordinators Juan Ruben and Susana Navarro Ordonez).
Conflict-of-interest disclosure
Lydie M. Da Costa: no competing financial interests to declare.
Isabelle Marie: no competing financial interests to declare.
Thierry M. Leblanc: no competing financial interests to declare.
Off-label drug use
Lydie M. Da Costa: nothing to disclose.
Isabelle Marie: nothing to disclose.
Thierry M. Leblanc: nothing to disclose.