Abstract
Severe immune cytopenias (SICs) are rare acquired conditions characterized by immune-mediated blood cell destruction. They may necessitate emergency medical management and long-term immunosuppressive therapy, strongly compromising the quality of life. The initial diagnostic workup involves excluding malignancies, congenital cytopenias, bone marrow failure syndromes, infections, and rheumatologic diseases such as systemic lupus erythematosus. Causal factors for SIC such as primary immunodeficiencies or immune regulatory disorders, which are referred to as inborn errors of immunity (IEIs), should be diagnosed as early as possible to allow the initiation of a targeted therapy and avoid multiple lines of ineffective treatment. Ideally, this therapy is directed against an overexpressed or overactive gene product or substitutes a defective protein, restoring the impaired pathway; it can also act indirectly, enhancing a countermechanism against the disease-causing defect. Ultimately, the diagnosis of an underling IEI in patients with refractory SIC may lead to evaluation for hematopoietic stem cell transplantation or gene therapy as a definitive treatment. Interdisciplinary care is highly recommended in this complex patient cohort. This case-based educational review supports decision making for patients with immune-mediated cytopenias and suspected inborn errors of immunity.
Learning Objectives
Severe immune cytopenia, eg, Evans syndrome, autoimmune hemolytic anemia, or immune thrombocytopenia, is one possible, early, potentially dangerous, and difficult-to-treat manifestation of an underlying inborn error of immunity (IEI)
Physicians must diagnose an underlying IEI to identify an effective treatment strategy for immune-mediated cytopenia
In some cases, physicians may choose a precise, existing therapy that directly targets the pathomechanism of the IEI; in other cases, they may choose a semitargeted approach based on the category of the IEI, which may allow them to effectively adapt cytopenia-directed treatment algorithms
Refractory or recurring cytopenia may serve as an additional indication, encouraging physicians to evaluate a patient with IEI for gene therapy or hematopoietic stem cell transplantation
Clinical case: part 1 of 3
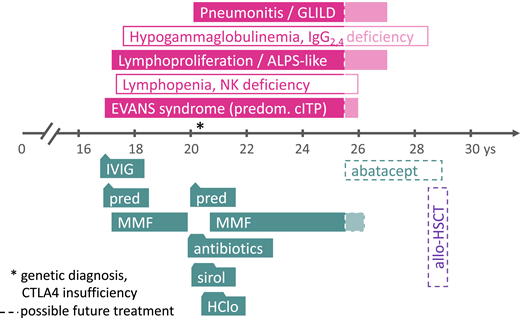
A 17-year-old boy presented with epistaxis that necessitated tamponade and severe thrombocytopenia (5 g/L; Figure 1). Furthermore, the results of laboratory investigations showed severe neutropenia (0.4 g/L) and a positive Coombs test (1:16), normal hemoglobin, reticulocytes, and red blood cell counts. These results led physicians to suspect Evans syndrome (ES) with predominant immune thrombocytopenia and neutropenia. Because the administration of high-dose intravenous immunoglobulin (IVIG, 5 days of 0.5 g/kg/d) improved the platelet counts only transiently and moderately but did not affect absolute neutrophil counts, high-dose prednisolone was started on day 8. This regimen was successful, as platelet counts rose to around 50 g/L and the absolute neutrophil count normalized; mycophenolate mofetil (MMF) was then added on day 17 as a glucocorticosteroid-sparing measure. No clinical signs of immunodeficiency or a history suggestive of an inborn error of immunity (IEI) were present. The prednisolone dosage was slowly reduced after the full dose (2 to 3 mg/kg/d) had been given for 4 weeks; this could be terminated around week 9 after the first presentation, rendering MMF as monotherapy. At that time, no attempts were made to perform a genetic diagnosis, although the complete absence of CD56+ natural killer cells and moderate lymphopenia (0.7 to 1 g/L, normal relative distribution of CD19+ B and CD4+ and CD8+ T cells) was noted. The total immunoglobulin (IgG, IgA, IgM) concentrations and specific antibody production amounts were normal.
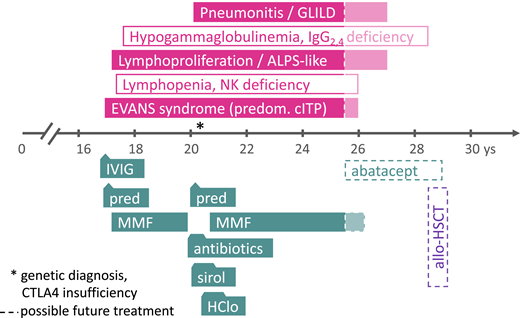
Clinical case presentation. The symptoms and disease phenotypes (in filled boxes) and predominant laboratory abnormalities (in empty boxes) are depicted in the upper panel, the time axis shows the patient’s age in years (ys). Treatment phases with various agents are shown in the lower panel (boxes with triangles or trapezoids to indicate shorter durations of treatment courses than the boxes suggest). The dashed lines and shaded area on the right side (from 25 years of age and onward) indicate future treatment options; the asterisk indicates the time point of the genetic diagnosis. allo-HSCT, allogeneic hematopoietic stem cell transplantation; ALPS, autoimmune lymphoproliferative syndrome; GLILD, granulomatous lymphocytic interstitial pneumonitis; HClo, hydroxychloroquine; IgG, immunoglobulin G; IVIG, high-dose intravenous immunoglobulin G; MMF, mycophenolate mofetil; NK, natural killer cells; pred, prednisolone; predom. cITP, predominantly chronic immune thrombocytopenia (as part of multilineage autoimmune cytopenia); sirol, sirolimus.
Clinical case presentation. The symptoms and disease phenotypes (in filled boxes) and predominant laboratory abnormalities (in empty boxes) are depicted in the upper panel, the time axis shows the patient’s age in years (ys). Treatment phases with various agents are shown in the lower panel (boxes with triangles or trapezoids to indicate shorter durations of treatment courses than the boxes suggest). The dashed lines and shaded area on the right side (from 25 years of age and onward) indicate future treatment options; the asterisk indicates the time point of the genetic diagnosis. allo-HSCT, allogeneic hematopoietic stem cell transplantation; ALPS, autoimmune lymphoproliferative syndrome; GLILD, granulomatous lymphocytic interstitial pneumonitis; HClo, hydroxychloroquine; IgG, immunoglobulin G; IVIG, high-dose intravenous immunoglobulin G; MMF, mycophenolate mofetil; NK, natural killer cells; pred, prednisolone; predom. cITP, predominantly chronic immune thrombocytopenia (as part of multilineage autoimmune cytopenia); sirol, sirolimus.
Treatment options for immune cytopenia in patients with IEIs
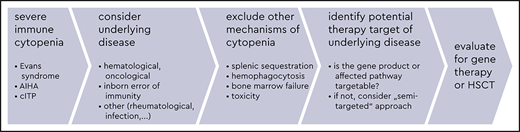
In general, first-line therapy for severe immune cytopenia (SIC) in patients with suspected IEIs follows established guidelines for the respective “primary” condition, that is, the immune cytopenia without a known underlying disease (Figure 2, left panel, and Table 1).1-5 However, an improvement in efficacy may be expected if an underlying condition (such as the IEI) is identified and can be treated simultaneously (Figure 2, right panel).6-9 For some IEIs, a targeted therapy can compensate for the underlying defect, restoring the impaired signaling pathway and potentially correcting the accompanying autoimmune cytopenia. In other, more unspecific IEIs, defining at least the category of IEI (eg, whether it is a combined immunodeficiency, a predominantly antibody deficiency, or an autoinflammatory syndrome) can help the physician choose a second-line therapy that is at least directed towards the suspected pathomechanism.
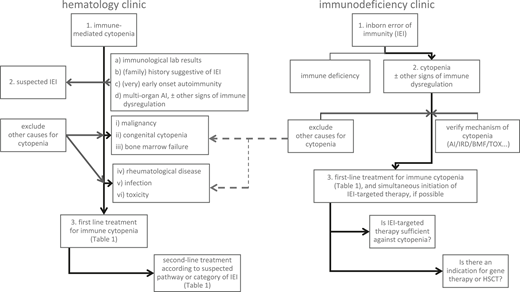
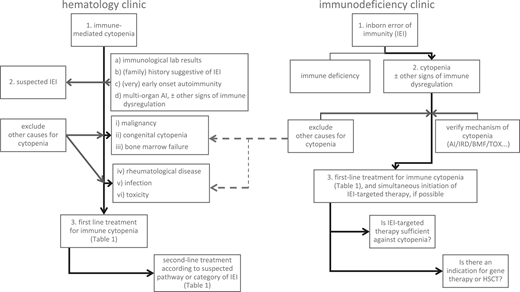
Management algorithm: scenarios of a patient with immune-mediated cytopenia at the hematology clinic without known IEI (left panel) or with a previously known IEI and manifestation of cytopenia at the immunodeficiency clinic (right panel). AI, autoimmune; BMF, bone marrow failure; HSCT, allogeneic hematopoietic stem cell transplantation; IEI, inborn error of immunity; IRD, immune regulatory disorder/immune dysregulation; TOX, infection- or drug-mediated myelotoxicity.
Management algorithm: scenarios of a patient with immune-mediated cytopenia at the hematology clinic without known IEI (left panel) or with a previously known IEI and manifestation of cytopenia at the immunodeficiency clinic (right panel). AI, autoimmune; BMF, bone marrow failure; HSCT, allogeneic hematopoietic stem cell transplantation; IEI, inborn error of immunity; IRD, immune regulatory disorder/immune dysregulation; TOX, infection- or drug-mediated myelotoxicity.
Furthermore, the treatment goal must be defined: In ES and warm autoimmune hemolytic anemia (AIHA), the international standard of care is to aim at the induction of remission as soon as possible, whereas the primary aim in chronic immune thrombocytopenia is to reduce the risk of bleeding while not compromising the quality of life. In conditions with hypersplenism, structural cytopenia, or bone marrow failure and simultaneous autoimmunity (eg, thrombocytopenia in Wiskott–Aldrich syndrome or immune thrombocytopenia in Fanconi anemia10 ) it might be difficult to assess the relative contribution of each cause of cytopenia. The avoidance of irreversible, iatrogenic damage is of utmost importance in these complex situations.
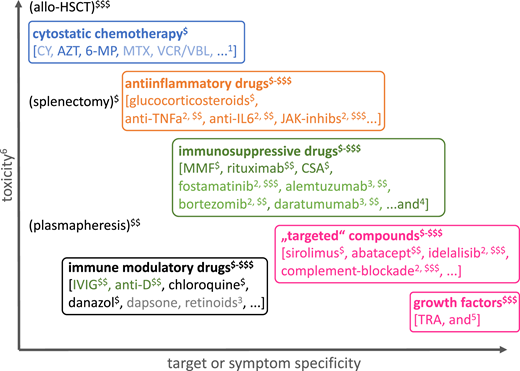
Finally, physicians must consider factors other than the efficacy and target or mechanism specificity of the treatment, including assessing the off-target toxicity (eg, teratogenicity, organ toxicity, additional pharmacologic immunosuppression, reversibility) against the treatment efficacy and costs. Figure 3 presents the various categories of drugs and other therapeutic measures used in cytopenias in IEI against this background.
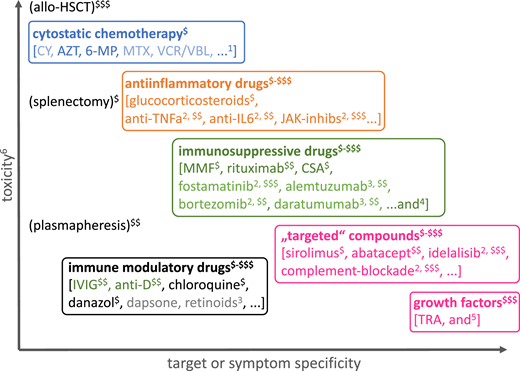
Classification of drugs used for the treatment of immune-mediated cytopenia according to their mechanism of action, toxicity, target, or symptom specificity and treatment costs. The large overlap of some of these subgroups (especially of anti-inflammatory drugs, immunosuppressive drugs, “targeted” drugs) is not shown for clarity. 6-MP, 6-mercaptopurine; anti-D, anti-Rh(D) antibody; AZT, azathioprine; CSA, cyclosporin A; CY, cyclophosphamide; IVIG, high-dose intravenous immunoglobulin G; JAK-inhibs, inhibitors of Janus kinases; MMF, mycophenolate mofetil; MTX, methotrexate; TNFa, tumor necrosis factor α; TRA, thrombopoietin receptor agonists; VCR/VBL, vinca alkaloids. 1Rarely used; etoposide in hemophagocytic lymphohistiocytosis; 2use in strictly defined indications; 3use based on largely anecdotal evidence, ideally under clinical trial conditions or compassionate use with informed consent; 4ibrutinib (targeting BTK, ITK), belimumab (anti-BAFF), epratuzumab (anti-CD22), carfilzomib (proteasome inhibitor); 5G-CSF in severe aplastic anemia, CXCR4 inhibition in myelokathexis, rarely erythropoietin analogs; 6the overall toxicity is difficult to measure, taking teratogenicity, myelo- and organ toxicity, immunosuppression, reversibility, and procedural risks into account, and is schematically illustrated here without considering individual risk factors (intolerance and acquired or inherited risk factors, eg, for thromboembolism or allergic reactions) or potential specific adverse effects; $-$$$a very rough estimate of procurement and treatment costs over repeated or continuous application is presented from “budget” ($, ≤US$1500/year) to high ($$$, ≥US$15 000-20 000/year).
Classification of drugs used for the treatment of immune-mediated cytopenia according to their mechanism of action, toxicity, target, or symptom specificity and treatment costs. The large overlap of some of these subgroups (especially of anti-inflammatory drugs, immunosuppressive drugs, “targeted” drugs) is not shown for clarity. 6-MP, 6-mercaptopurine; anti-D, anti-Rh(D) antibody; AZT, azathioprine; CSA, cyclosporin A; CY, cyclophosphamide; IVIG, high-dose intravenous immunoglobulin G; JAK-inhibs, inhibitors of Janus kinases; MMF, mycophenolate mofetil; MTX, methotrexate; TNFa, tumor necrosis factor α; TRA, thrombopoietin receptor agonists; VCR/VBL, vinca alkaloids. 1Rarely used; etoposide in hemophagocytic lymphohistiocytosis; 2use in strictly defined indications; 3use based on largely anecdotal evidence, ideally under clinical trial conditions or compassionate use with informed consent; 4ibrutinib (targeting BTK, ITK), belimumab (anti-BAFF), epratuzumab (anti-CD22), carfilzomib (proteasome inhibitor); 5G-CSF in severe aplastic anemia, CXCR4 inhibition in myelokathexis, rarely erythropoietin analogs; 6the overall toxicity is difficult to measure, taking teratogenicity, myelo- and organ toxicity, immunosuppression, reversibility, and procedural risks into account, and is schematically illustrated here without considering individual risk factors (intolerance and acquired or inherited risk factors, eg, for thromboembolism or allergic reactions) or potential specific adverse effects; $-$$$a very rough estimate of procurement and treatment costs over repeated or continuous application is presented from “budget” ($, ≤US$1500/year) to high ($$$, ≥US$15 000-20 000/year).
Clinical case: part 2 of 3
Results of more in-depth immunologic analyses at later time points showed a borderline increased proportion of T-cell receptor α-β-positive CD4- and CD8-negative (double negative) T cells (DNT, 4% to 8% of CD3+; normal <2.5). The disease course was complicated by an episode of severe respiratory problems (chest pain and respiratory distress), which initially led to polypragmatic treatment with antibacterial and antifungal antibiotics and a pause in MMF provision. Ultimately, a biopsy with subsequent histopathological assessment led to the additional diagnosis of granulomatous lymphocytic interstitial lung disease (GLILD; Figure 1). Symptoms and radiographic pathologies improved, but ES recurred; because MMF had been previously well tolerated, it could be successfully reintroduced.
Approach to the treatment decision for SIC in patients with IEIs
Multiple scenarios arise in real life that influence early treatment decisions in this patient cohort (Figure 2).
Scenario 1: a patient with immune cytopenia at the hematology clinic
An underlying IEI is unknown but possible because of either:
Abnormal immunological laboratory results
Family history of immunodeficiency (infections) or immune dysregulation (autoimmunity, autoinflammation, etc.)
Very early onset, relapsing or refractory course
Multiorgan autoimmunity or immune dysregulation.
A physician treating any patient with a so-called primary SIC seen at a hematology clinic should subsequently exclude underlying causes in order to more precisely manage the condition. For patients with suspected IEI, various possible pathomechanisms can cause cytopenia, all of which have to be considered when selecting the treatment.6,8 Autoimmunity in the strict sense, whether it is caused by a primary B-cell pathology, as is seen in common variable immunodeficiency (CVID), or a T-cell maturation/differentiation or functional impairment, as is seen in combined immunodeficiencies (CIDs) or primary immune regulatory disorders (PIRDs), all can result in the failure of tolerance mechanisms and autoreactivity and are usually associated with detectable autoantibodies against blood cells. However, many other features of immune dysregulation may be present in patients with IEI. These other features include hemophagocytosis, lymphoproliferation with subsequent splenic sequestration of blood cells, chronic autoinflammation or complement hyperactivation (eg, in defects of phagocytes or of intrinsic and innate immunity, autoinflammatory syndromes, or complement deficiencies), bone marrow failure, or myelotoxicity in the context of infections or drug therapies.6,11,12
If hematologists note the presence of any of the aforementioned risk factors (1-4; Figure 2, left panel, a-d) in a patient with immune cytopenia, they should suspect the existence of an underlying disorder, thus a “secondary” immune cytopenia, and initiate an earlier immunologic and genetic evaluation than in the general population with no detectable underlying cause for immune cytopenia (Figure 2, left panel). GLILD, as detected in the presented patient during the progress of his disease and treatment course, is one of the major, seriously compromising autoimmune and autoinflammatory manifestations in patients with certain IEIs such as CID or CVID.13 The hematologist is well advised to consult or involve an immunologist at this point. Likewise, if the regular first-line treatment that would be provided to meet international and local standards is unsuccessful for such a patient, a rapid escalation to second-line treatment, potentially targeting IEI-linked pathomechanisms, is recommended (Table 1; Figure 2, left panel).1,3-5,8
Although making a definitive diagnosis of the suspected underlying IEI should be assigned first priority, it could take months until the specific IEI is identified. Meanwhile, a semitargeted treatment that is guided by immune phenotypical clinical and laboratory parameters can be initiated before this diagnosis is achieved. For instance, if the hematologist notes signs of predominant antibody deficiency and impaired B-cell maturation and subset distribution, a decision to choose a B-cell–directed strategy might be more rapidly made. Alternatively, if the hematologist detects an increased proportion of DNT cells, sirolimus and MMF might be considered as preferred therapies (Table 1). Many IEIs that may manifest with autoimmune cytopenias share some immune phenotypical abnormalities, such as a phenotype of exhaustion and senescence in certain lymphocyte subsets,14-17 but they may be distinguished from one another by specific parameters, even before a genetic diagnosis can be made. A prospective study is ongoing in which researchers strive to identify biomarkers that predict treatment responses among patients with these immediately available immune phenotypical abnormalities and analyze their transcriptome and epigenetic modifiers (www.sic-reg.org).18
Clinical case: part 3 of 3
Repeated attempts to reduce the MMF dosage over the years of the patient’s disease course were unsuccessful. A genetic analysis that became available later in the patient’s course revealed a heterozygous pathogenic variant in CTLA4 (c.2223C>T; p.R75W; ENST00000302823). On the basis of this (in the meantime known) CTLA4 haploinsufficiency, a “semitargeted” treatment attempt with hydroxychloroquine and sirolimus was made, omitting MMF temporarily (Figure 1). However, because pancytopenia recurred, MMF was reinitiated, again being successful as monotherapy. Finally, the patient was transitioned to adult hematology with mild to moderate thrombocytopenia and good partial remission from GLILD (only small residual opacities on chest computed tomography without lung function impairment) while still receiving low-dose MMF treatment (15 mg/kg/d). In future situations of deterioration, a targeted treatment using abatacept would be indicated, and in severe cases of CTLA4 insufficiency, even hematopoietic stem cell transplantation (HSCT) remains an option (Figure 1, dashed/shaded boxes).19
Scenario 2: a patient with cytopenia at the immunodeficiency clinic
A defined IEI is known:
With a targetable pathway (many monogenic IEIs, eg, CTLA4 insufficiency)
Without a targetable pathway (eg, CVID not further specified)
If a monogenic IEI is known (a), the therapeutic priority should be to treat the underlying disease (Figure 2, right panel). However, autoimmune cytopenia may present as an emergency situation, and not every targeted drug acts immediately; a preparative treatment of eventual HSCT also does not cure autoimmune cytopenia effectively and rapidly. Therefore, the sequence of treatment steps and bridging strategies play important roles for cytopenias in IEI. The treating immunologist should consider involving hematologists as soon as possible so they can work in close cooperation. For instance, in LRBA deficiency, abatacept may be a highly effective treatment of many autoimmune phenomena, restoring the regulatory T-cell defect in part, but the onset of its effect can be delayed for days or weeks. Thus, a short course of glucocorticoids or other first-line treatment measures may be needed to treat AIHA or ES in this context, even if a targeted drug is available (Table 1). Similarly, MMF or sirolimus may be considered as steroid-sparing therapies, for which time is needed to reach effective plasma concentrations and exploit their full therapeutic potential.
In certain situations, targeted drugs only ameliorate some of the features of an IEI, and cytopenia may arise as new manifestation in a patient despite the fact that he or she is already receiving targeted treatment. Physicians should choose drug combinations that target both the underlying pathway and the suspected mechanism of the immune-mediated cytopenia on an individual basis (eg, a combination of abatacept with sirolimus for a patient with LRBA deficiency). Likewise, a study of patients with a transplant indication with IEI and immune dysregulation showed that those who displayed reduced disease activity before HSCT had significantly better outcomes.20 This reduction in disease activity can be achieved by inducing at least a partial remission with targeted anti-inflammatory and immunosuppressive pretreatment.20 If the molecular target or impaired signaling pathway is unknown (b), categorizing the primary immunodeficiency according to clinical and laboratory immune phenotype parameters and classifications11,21,22 may help guide treatment decisions, as delineated in Table 1. Close cooperation between the immunologist and the hematologist is recommended.
Scenario 3: reevaluation of a patient with recurring cytopenia in the hematology or immunology clinic
The patient had received lines of treatment previously and had mixed responses.
Which earlier therapies were partially successful? Were the algorithms for the treatment of immune cytopenias in the general population followed?
Is there any newly detectable parameter that points toward a possible treatment target (eg, DNT cells, hypogammaglobulinemia, naive T and B cells, exhaustion and senescence markers; see Table 1)?
Is there any historical, clinical, or earlier laboratory parameter that points toward a different differential diagnosis other than an autoimmune condition (eg, congenital cytopenia, bone marrow failure or malignancy, splenic sequestration, hemophagocytosis, chronic infection, toxic drug exposure)? Was the evolution of bone marrow failure or of a clonal disease excluded?
The combination of a refractory nature of autoimmune cytopenia and severe additional immunological phenotypes or risk of malignancy (if inherent in the respective IEI) should prompt consideration of evaluating for HSCT.
In this scenario, the expert immunologist is already cooperating effectively with the expert hematologist to provide patient care. It is always warranted to repeat the evaluation and perform a thorough differential diagnostic workup to exclude other causes for patients with autoimmune cytopenias, especially in refractory or frequently recurring situations and where a monogenic cause cannot be identified or has been insufficiently described. For example, many IEIs do not exclude but rather increase the risk of malignancy or bone marrow failure as an underlying cause of cytopenia, such as in a deficiency of CTLA4, ADA2, GATA2, or dyskeratosis congenita.23-28 Furthermore, and independent of his or her IEI, a patient with CVID may suffer from an additional trait linked to congenital, mild thrombocytopenia that is aggravated by CVID-linked splenomegaly. Therefore, composite mechanisms that contribute to the manifestation and extent of cytopenia must be considered for patients with IEI; this can ideally be achieved by a close cooperation between hematologists and immunologists.
Scenario 4: autoimmune cytopenia newly arising in a patient at the transplant clinic or ward
Autoimmune cytopenias for patients with secondary immunodeficiency, such as after allogeneic HSCT or after organ transplantation, are among the most difficult to diagnose and treat. Recent studies and reviews that address this topic specifically29-31 provide more in-depth guidelines than this present work focusing on SIC in primary immunodeficiencies. In brief, autoimmune cytopenias in secondary immunodeficiencies such as post-transplantation settings may be caused by a complex mix of underlying pathomechanisms such as imbalanced immune reconstitution with preponderance or insufficient elimination of autoreactive T- or B-cell clones, calcineurin inhibitor–facilitated skewing of the immune system, drug-induced thrombotic microangiopathy or sinusoidal obstruction syndrome, and virus-triggered conventional autoimmune reactions, to list a few. Their manifestation probably will warrant a change of the post-transplant pharmacologic immunosuppressive treatment regimen and soon need escalation toward second- and third-line therapies, including even unconventional measures (eg, combinations of anti-T and anti-B cell–directed interventions, complement inhibition). Of note, mTOR inhibition might be a favorable alternative immunosuppression to switch to in this condition, as the enhancement of regulatory T-cell function was demonstrated under rapamycin.32
Conclusions
Immune cytopenias may follow a more complicated course than seen in the general population if they occur in patients with an underlying IEI. Like other features of immune dysregulation, the manifestation of immune cytopenia may precede an increased frequency or severity of infections, actually impeding the early diagnosis of an IEI. These scenarios highlight the explicit reasons hematologists and immunologists should jointly care for this selected patient cohort from the beginning of treatment and onward. Targeted, precise treatment options may exist for selected IEIs, and these options have the potential to cure or control autoimmune cytopenia and reduce off-target adverse effects. To support decision making in the future, prospective studies to define treatment response–predicting or –stratifying biomarkers for patients with autoimmune cytopenias and IEI are needed. Ultimately, a relapsing or refractory course of autoimmune cytopenia observed in patients with an IEI may represent a major indication to proceed to gene therapy or HSCT.
Contributions: M.G.S. wrote the manuscript and designed figures and tables.
1Rarely used; etoposide in hemophagocytic lymphohistiocytosis.
2Use in strictly defined indications.
3Use based largely on anecdotal evidence, ideally under clinical trial conditions or compassionate use with informed consent.
4Ibrutinib (targeting BTK, ITK), belimumab (anti-BAFF), epratuzumab (anti-CD22), carfilzomib (proteasome inhibitor).
5Granulocyte colony-stimulating factor in severe aplastic anemia, CXCR4 inhibition in myelokathexis, rarely erythropoietin analogues.
6The overall toxicity is difficult to measure, taking teratogenicity, myelotoxicity and organ toxicity, immunosuppression, reversibility, and procedural risks into account, and is schematically illustrated here without considering individual risk factors (intolerance, acquired or inherited risk factors, eg for thromboembolism or allergic reactions) or potential specific adverse effects.
$–$$$A very rough estimate of procurement and treatment costs over repeated or continuous application is presented from “budget” ($ ≤1,500 USD/year) to high ($$$ ≥15,000–20,000 USD/year).
Acknowledgments
Sara Crockett (Graz) is thanked for scientific editing. M.G.S. and the research unit for pediatric hematology and immunology are supported in part by the Styrian Children’s Cancer Aid (Steirische Kinderkrebshilfe) Foundation.