Abstract
The identification of genetic disorders associated with dysregulated immunity has upended the notion that germline pathogenic variants in immune genes universally result in susceptibility to infection. Immune dysregulation (autoimmunity, autoinflammation, lymphoproliferation, and malignancy) and immunodeficiency (susceptibility to infection) represent 2 sides of the same coin and are not mutually exclusive. Also, although autoimmunity implies dysregulation within the adaptive immune system and autoinflammation indicates disordered innate immunity, these lines may be blurred, depending on the genetic defect and diversity in clinical and immunological phenotypes. Patients with immune dysregulatory disorders may present to a variety of clinical specialties, depending on the dominant clinical features. Therefore, awareness of these disorders, which may manifest at any age, is essential to avoid a protracted diagnostic evaluation and associated complications. Availability of and access to expanded immunological testing has altered the diagnostic landscape for immunological diseases. Nonetheless, there are constraints in using these resources due to a lack of awareness, challenges in systematic and logical evaluation, interpretation of results, and using results to justify additional advanced testing, when needed. The ability to molecularly characterize immune defects and develop “bespoke” therapy and management mandates a new paradigm for diagnostic evaluation of these patients. The immunological tests run the gamut from triage to confirmation and can be used for both diagnosis and refinement of treatment or management strategies. However, the complexity of testing and interpretation of results often necessitates dialogue between laboratory immunologists and specialty physicians to ensure timely and appropriate use of testing and delivery of care.
Learning Objectives
Describe the phenotypic manifestations and molecular mechanism of CTLA-4 haploinsufficiency
Discuss the approach to diagnostic evaluation of patients with autoimmune cytopenias in the context of inborn errors of immunity
Introduction
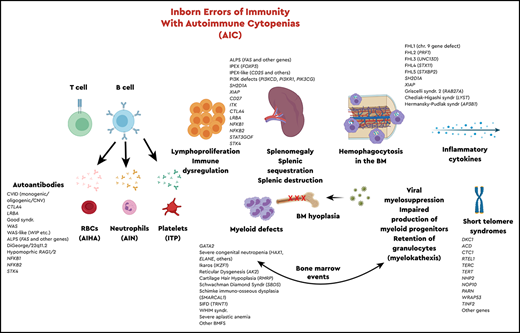
The most recent classification of inborn errors of immunity (IEIs)1 included a total of 416 genes, 45 of which are associated with immune dysregulation2 and 43 of which are associated with bone marrow failure syndromes, based on clinical phenotypes and inheritance patterns. In the last 6 months, since publication of the most recent classification, ∼30 new monogenic IEIs have been reported. Abnormal immune regulation and persistent inflammation are hallmarks of autoimmune disease, and hematological anomalies affecting one or more cell lines are common manifestations of autoimmunity and often reflect underlying disease activity. There is also a relationship between chronic immune dysregulation and lymphoproliferative disease, likely related to persistent stimulation, and subsequent proliferative response. It is essential for hematologists and immunologists to recognize and understand the intersection of immunology and hematology, especially with regard to genetic disorders of the immune system that predispose to hematological autoimmunity, benign or malignant lymphoproliferation, and bone marrow failure.
This article uses a case-based approach to review and highlight a specific genetic inborn error as an exemplar of immune dysregulation that results in multiple autoimmunity with hematological aberrations, lymphoproliferation, susceptibility to malignancies, and end-organ damage. This article focuses primarily on the laboratory diagnosis and work-up for suspected autoimmune cytopenias (AICs) in the context of monogenic immune dysregulation.
Clinical case
A 42-year-old woman with a 15-year history presented with progressive headache and neck pain, stiffness, and reduced hearing, with no fever or respiratory symptoms. Her symptoms of headache progressed with vomiting and horizontal diplopia, and imaging (computed tomography, magnetic resonance imaging) revealed an enhancing anomaly in the brain parenchyma. The finding of pathogen testing was negative, and treatment with IV steroids resulted in symptom resolution. About 8 months after the initial episode of the above-mentioned neurological symptoms, she developed a similar presentation and was diagnosed with multiple sclerosis, and she was initiated on immunomodulatory therapy. Approximately 4 years from the initial presentation, she was noted to be mildly anemic, and thoracic imaging revealed mediastinal and axillary adenopathy as well as splenomegaly. Multiple lymph node biopsies only revealed lymphoid hyperplasia. She later developed jaundice with extreme fatigue and shortness of breath. A complete blood count revealed a hemoglobin of 4 g/dL, and a diagnosis of hemolytic anemia was made. A few months later, she developed thrombocytopenia and shingles involving a thoracic dermatome. Her platelet count dropped to 1000 per microliter, requiring platelet transfusions. The cytopenias were treated with steroids, immunoglobulin, and rituximab. There appeared to be progression of disease over time, and the brain lesions were considered as part of an undefined lymphoproliferative process. The hematologist suggested an allogeneic hematopoietic cell transplant (HCT), despite the lack of a clear and unifying clinical diagnosis. The key questions at this point are the likely differential diagnoses and which laboratory evaluation to use to arrive at a diagnosis.
Approach to diagnostic evaluation of the clinical case
The clinical phenotype of the patient suggests an immune dysregulatory disorder. Laboratory evaluation for autoimmune lymphoproliferative syndrome (ALPS) was performed because of her autoimmune hemolytic anemia (AIHA), thrombocytopenia (presumed autoimmune), and lymphoproliferation. Flow cytometric analysis to assess for increased T-cell receptor α-β+ double-negative (CD3+CD4−CD8−) T cells revealed they were within normal limits and not expanded, as would be seen with typical ALPS.3-5 Her immunoglobulin levels for immunoglobulin G (IgG), IgA, and IgM revealed modest hypogammaglobulinemia with partial IgA deficiency. She had intact vaccine antibody responses to protein-based vaccines but abnormal antibody responses to pneumococcal polysaccharide vaccination.
On the basis of these findings, she was given a presumptive diagnosis of common variable immunodeficiency (CVID), which was supported by her clinical findings of interstitial lung disease and AICs. Detailed flow cytometric analysis of peripheral blood B-cell subsets revealed significant defects in B-cell differentiation with decreased total memory and class-switched memory B cells (0.1%). She appeared to have recovered total peripheral B-cell counts after multiple treatments with rituximab, but she did not have normal B-cell differentiation. She also had decreased plasmablasts with substantially increased CD21-negative B cells (marker of immature B cells). At the time of this evaluation, targeted gene sequencing panels and whole-exome sequencing (WES) were not common. CVID does not represent a single diagnostic entity, but rather encompasses several distinct monogenic defects, and the majority of patients may have an oligogenic or polygenic etiology. Several years later, as genetic testing became increasingly available and affordable, she underwent a targeted next-generation sequencing (NGS) panel for IEIs, which reported a likely pathogenic heterozygous variant in the CTLA4 gene, c.410C>T, p.Pro137Leu, consistent with CTLA-4 haploinsufficiency. CTLA-4 haploinsufficiency with autoimmunity (CHAI) is an IEI resulting from loss of a key immunoregulatory molecule that functions as a “checkpoint” inhibitor of T-cell activation.
Understanding the predisposition to autoimmunity in the context of CTLA-4 deficiency
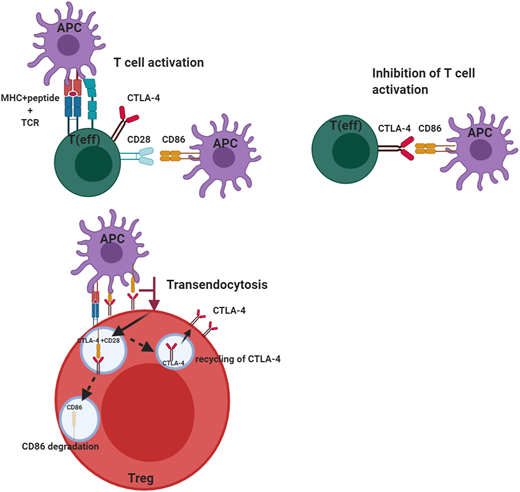
The cellular adaptive immune system (ie, T cells) uses several regulators to ensure there is an appropriate counterbalance to T-cell activation to prevent uncontrolled T-cell activation. Among these is CTLA-4, a key negative regulator of the peripheral immune response (Figure 1). CTLA-4 is expressed by CD4+ and CD8+ T cells and is only expressed on activated T cells, but it is constitutively present on regulatory T cells (Tregs). CTLA-4, like the constitutive T-cell costimulation molecule CD28, binds the ligands B7-1 (CD80) and B7-2 (CD86). The difference is CTLA-4 binds these molecules with much higher affinity and avidity than CD28, enabling it to serve as a “brake” to the T-cell activation response. In the case of Tregs, the small amounts of CTLA-4, which are constitutively expressed, are rapidly upregulated on activation of T cells. Although the immunosuppressive effects of Tregs are mediated through a variety of mechanisms, including immunomodulation via CTLA-4, the absence of this molecule affects both Treg numbers and function. This may be due to CTLA-4 being a target gene of FOXP3, which is a critical regulator of Tregs and is required for the effective suppressor function of these cells. Because CTLA-4 is typically sequestered in the intracellular compartment and substantially upregulated upon T-cell activation, it has to capture its ligands, B7-1 and B7-2, which it does via a process called transendocytosis, allowing these molecules to be degraded inside the cells, which express CTLA-4. This process requires sufficient CTLA-4 expression to be present on the surface of Tregs or activated T cells because the ability to remove the ligands is dependent both on cell contact and the duration of cell contact. Once CTLA-4 is internalized, it is either degraded in the lysosome or recycled to the plasma membrane. Another molecule, LPS-responsive beige-like anchor (LRBA), is colocalized in the endosome and recycles CTLA-4, allowing it to be reexpressed on the cell surface. This is why LRBA deficiency represents the “other side of the coin” with a clinical phenotype similar to CTLA-4 haploinsufficiency, especially in the predisposition to autoimmunity.6
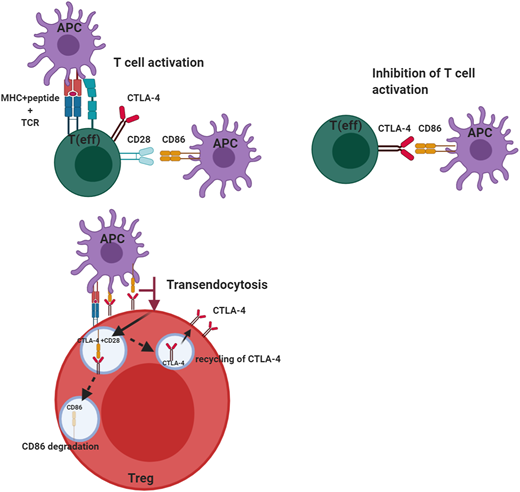
Role of CTLA-4 in the regulation of effector T-cell and regulatory T-cell function. CTLA-4 serves to inhibit T-cell activation in effector T cells after CD28 has interacted with its ligands B7-1 (CD80) and B7-2 (CD86). This inhibition by CTLA-4 prevents unbridled T-cell activation. CTLA-4 expressed on regulatory T cells can transendocytose the ligand CD86 by removing it from the antigen-presenting cell and internalizing it, after which the ligand is degraded and CTLA-4 is recycled to the cell membrane. This transendocytosis process is exploited in the assessment of CTLA-4 variants. This figure was created with BioRender.com.
Role of CTLA-4 in the regulation of effector T-cell and regulatory T-cell function. CTLA-4 serves to inhibit T-cell activation in effector T cells after CD28 has interacted with its ligands B7-1 (CD80) and B7-2 (CD86). This inhibition by CTLA-4 prevents unbridled T-cell activation. CTLA-4 expressed on regulatory T cells can transendocytose the ligand CD86 by removing it from the antigen-presenting cell and internalizing it, after which the ligand is degraded and CTLA-4 is recycled to the cell membrane. This transendocytosis process is exploited in the assessment of CTLA-4 variants. This figure was created with BioRender.com.
The clinical significance of ineffective CTLA-4 function has been amply demonstrated through patients who have heterozygous (autosomal dominant) pathogenic variants in CTLA4 resulting in haploinsufficiency.6-11 The presence of multiple autoimmunity in these patients, including AICs, enteropathy, type 1 diabetes, and neurological phenotypes, all indicate that insufficient CTLA-4 protein expression (caused by haploinsufficiency) impedes effective suppressive function of Tregs and leads to inadequate control of T-cell activation. These patients also often demonstrate decreased immunoglobulins (immunodeficiency with predisposition to respiratory infections) and nonmalignant lymphoproliferation. The inflammatory phenotype in CTLA-4 haploinsufficiency does not necessarily correlate with lymphocyte expansion and infiltration and is likely dependent on the genetic variant.12
Laboratory diagnosis and work-up for AICs in the context of immune dysregulatory disorders
Serological and immunophenotyping assessment
There is a growing list of single-gene defects associated with immune dysregulation and manifesting with multiple hematological and immunological phenotypes, including AICs. Therefore, it is important to know when and how to evaluate a patient with AIC for an underlying immune dysregulatory disorder, to differentiate between monogenic and polygenic causes of disease, and to recognize the implications for personalized therapy in patients with specific molecular defects. Timely laboratory evaluation in the context of clinical phenotype can reduce the diagnostic odyssey of many patients and may prevent institution of nonspecific immunomodulatory therapies, which may increase the risk of infection, while providing minimal therapeutic benefit.
It is helpful to do a preliminary serological and immunophenotyping assessment, either in parallel with obtaining genetic analysis or before it, because it is often useful to have an immunophenotype to correlate with genetic data. Because patients with monogenic immune dysregulation, also called primary immune regulatory disorders, may present with hypo- or hypergammaglobulinemia, depending on the underlying context, quantitative measurement of serum immunoglobulins, especially IgG, IgA, IgE, and IgM, is useful to contextualize the underlying defect. Other serological assessments for autoimmunity and/or inflammation can include evaluation of soluble (s) biomarkers, such as B-cell activating factor, sCD163 (soluble haptoglobin, a marker for macrophage activation), and sCD25, as well as autoantibodies directed against neutrophils, platelets, and erythrocytes, for the assessment of autoimmune neutropenia (AIN), thrombocytopenia (immune thrombocytopenic purpura [ITP]), and AIHA. Autoantibody testing for platelets and neutrophils is controversial and often of limited utility due to analytical constraints and the low sensitivity of many of the available assays. Therefore, although autoantibody testing is often ordered in clinical practice, a negative result is not necessarily clinically informative. A complete blood count is useful for serial monitoring of blood counts, especially in response to treatment. sCD25 is a biomarker that is significantly elevated in CTLA-4 and LRBA deficiencies.
Flow cytometry is a tremendously valuable tool in the diagnosis of IEIs and can be used for immunophenotyping, protein expression, and functional assessment.13 Lymphocyte subset quantitation of T, B, and natural killer (NK) cells in blood is useful in “global binning” of patients, based on whether they have numerical deficits in one or more lymphocyte subsets. However, additional immunophenotyping, particularly of the T-cell (CD4+ and CD8+) and B-cell compartments is also often necessary because there can be quantitative and functional dysregulation in differentiation of the adaptive immune system, which may not be apparent by a global lymphocyte subset analysis. This detailed immunophenotyping includes quantitation of naïve and memory T cells, activated T cells, senescent and exhausted T cells, follicular T helper cells, and expression of key cytotoxic proteins in NK cells and CD8+ T cells (perforin, granzymes) in the T-cell compartment (Table 1). Follicular T helper cells are substantially expanded in patients with CTLA-4 and LRBA defects. Among B-cell subsets, early (transitional and naïve B cells) and late B-cell (memory B cells, plasmablasts, CD21bright, and CD21dim) subsets should be quantitated in blood (Table 1). CD21dim (CD21low) B cells have been described in CVID as well as other secondary immune dysregulatory diseases, such as systemic lupus erythematosus, as an unusual naïve-like B cell, defective in B-cell receptor stimulation, and expanded in patients with AICs, splenomegaly, and granulomatous disease, which likely reflects the overall B-cell maturation defects in this group of immunological disorders.14-16 In addition, because autoimmunity is often associated with either quantitative or functional defects in Tregs, immunophenotyping of this compartment (CD4+CD25+/FOXP3+ T cells) is often considered useful for the laboratory evaluation of these conditions (Table 1). For example, in LRBA deficiency and CTLA haploinsufficiency, impairment in Treg function and quantitative reduction in Tregs, as previously noted, results in a strong predisposition to autoimmunity.6,17,18 Therefore, the ability to assess not only Treg numbers by flow cytometry but also the function of the suppressor cells is critical; however, functional Treg assays have not been validated in most clinical diagnostic laboratories and are thus mainly available only in a research setting. Furthermore, although there are several analytical approaches to assessing Treg function, not all of them are equally robust and reliable, and these assays continue to be explored and developed for validation in the clinical diagnostic laboratory.
Global functional evaluation of the immune system, not specific to a particular molecular defect, but broad interrogation of pathways (Table 1), includes T-cell proliferation to non-specific stimuli, specific antigens, and measurement of cytokine production among others. The non-specific mitogenic stimuli include phytohemagglutinin, phorbol myristate acetate/ionomycin, anti-CD3+/anti-CD28, anti-CD3+/interleukin-2 [IL-2], anti-CD3+/IL-7, while the specific antigens include Candida, tetanus toxoid or viral peptides to Epstein Barr virus, cytomegalovirus, Varicella zoster virus among others. Assessment of the T cell response could include production of cytokines, such as IL-2, interferon gamma and TNF-alpha, by CD4+ and CD8+ T cells, after stimulation with mitogens. It could also include cellular degranulation (CD107 expression) of cytotoxic lymphocytes (CD8+ T cells, NK cells), after appropriate specific or non-specific stimulation. Dysregulation of specific pathways, such as NF-κB or STAT, can be assessed by phosphorylation of key proteins after appropriate stimuli. Protein-specific expression in relevant cellular subsets, usually lymphocytes, can be determined for proteins when there is a high likelihood of loss of protein expression contributing to the clinical phenotype. It is helpful to include a functional assay, when possible, as an adjunct to a protein expression assay, especially for novel variants or atypical phenotypes. Assessment of various immunophenotypes, expansion or contraction, of specific T- or B-cell subsets, as described above, can also be useful in creating an immunophenotype to correlate with the clinical context and/or genetic data.
When considering diagnostic immunological testing, it is important to distinguish those assessed on a research basis vs validated testing performed in a clinical diagnostic laboratory. On the one hand, research-based testing is not widely available and therefore not easily accessible by most specialty clinicians in diverse practice settings. Clinical testing, on the other hand, is usually available to all clinicians, though there can be varying degrees of complexity in ordering such testing, depending on the nature of the practice.
Functional assessment of CTLA-4 and LRBA function
To directly assess the effect of CTLA-4 haploinsufficiency or LRBA deficiency, it is essential to have assays capable of assessing the specific molecules or pathways in question. Because CTLA-4 is upregulated on activated T cells within 24 to 48 hours after activation, and because its expression of Tregs is also increased after activation,19 flow cytometric measurement of CTLA-4 on the surface of activated T cells and Tregs provides one method of assessing if there is sufficient protein on the cell surface to mediate cellular suppressor function. However, the most definitive functional assay for these defects is the transendocytosis assay,20-22 which measures the removal of costimulatory ligands from antigen-presenting cells using flow cytometry and confocal microscopy. Inhibition of lysosomal degradation in this assay reveals an increase in the presence of B7-2 after transendocytosis (Figure 1). The ability of CTLA-4 to bind ligand and transendocytose is very dependent on the recycling process, which is mediated by LRBA, and thus, even if CTLA-4 expression is normal, if LRBA expression is impaired, it can affect ligand uptake. This allows a distinction between CTLA-4 and LRBA defects. Also, B7-1 has much higher affinity for CTLA-4 than does B7-2, and the ability to take up B7-2 is dependent on the presence of B7-1, but not vice versa.23 A combination of these assays is most useful, especially when characterizing novel CTLA4 and LRBA variants.
The role of genetic testing in identifying monogenic defects in patients with AICs
At the start of the third decade of the 21st century, it is undisputed that genetic evaluation plays a key role in the diagnosis of patients with IEIs, including those with autoimmune phenotypes, and this represents an ever-evolving and expanding area of study.24,25 Currently, the clinician is confronted with an embarrassment of riches when it comes to genetic testing because there are targeted NGS panels with defined sets of genes, WES, and whole-genome sequencing (WGS).26,27 Some key rules of thumb can be employed to determine which approach to use and to deploy these in a systematic and tiered manner. An American Academy of Allergy, Asthma, and Immunology committee has developed guidelines for selection of appropriate of genetic tests and interpretation of variants for IEIs.28 Currently, genetic testing is often done early in the diagnostic process when an IEI is suspected. However, although immunophenotyping and generic functional immune evaluation assays that are clinically available are often ordered almost immediately, genetic testing, depending on insurance and institutional regulations, may take a little longer. However, most patients who require genetic evaluation are usually able to obtain the testing with appropriate support from the physician, genetic counselor, and geneticist. More complex functional testing, either research based or clinical, is often required once the genetic result has been obtained, especially to further validate variants of uncertain significance (VUSs) or ambiguous results.
Targeted panels are most effective when there is a clear phenotype and/or family history, which supports the likelihood of a monogenic disease, and the panel under consideration is well represented for the genes of interest.29 The most recent International Union of Immunological Societies classification1 includes >400 genes, and most commercially available panels include most of these, which makes these panels practical and relatively rapid screens with a turnaround time averaging 2 or rarely 3 weeks. However, if the phenotype is more amorphous and ambiguous and there is either no family history or no samples for trio analysis (proband + parents) are available or there has been a previously negative targeted panel, WES is the most logical option.30 WES looks at a much larger sample of genes in an unbiased sequencing approach; however, it has its own limitations, including the type of bioinformatics pipeline that is used for variant calling and filtering. Clinical exomes that are available commercially are not all the same and can have variable coverage.31 Therefore, clinicians must be aware that not all exomes are created equal, and they need to understand the coverage and the type of bioinformatics approaches used, at least at a high level. A very recent analysis of the efficacy and cost of WES vs targeted panels revealed that, overall, going directly to a WES approach might be more cost effective in the long run, though diagnostic yield, depending on the cases, could be lower,32 indicating that an informed approach to selecting the method of genetic testing is required for each patient based on clinical and laboratory phenotype, family history, and other practical factors, including insurance coverage.
WGS has also been used for the evaluation of IEIs, but it is less common in the clinical setting as a first-tier test because of accessibility issues (payer restrictions unless other forms of genetic testing have been attempted) and complexities in data analysis and interpretation. Most often, WGS is employed for patients with complex phenotypes who have had a negative test result for either WES and/or targeted panels. WGS is particularly useful when disease may be caused by deep intronic variants (noncoding areas of the genome) or novel genes, which may not be well represented in clinical exomes.33,34 However, in patients who may have negative genetic testing but a clinical and/or laboratory phenotype strongly suggestive of monogenic disease, an iterative approach using reanalysis of WES data, WGS, or transcriptomic studies may prove useful in identifying a diagnosis.35 Transcriptomic studies, such as RNA sequencing, may be particularly useful in establishing the pathogenicity of variants in noncoding regions for rare disorders, such as IEIs.36,37 In addition, the relevance of closing the loop on genetic testing with functional studies and correlation with the clinical and immunological and other laboratory phenotype cannot be overemphasized and is useful not only for VUSs but also for known pathogenic variants, which may present with an atypical or expanded clinical phenotype (Figure 2). Reconfiguring the classification of VUSs by functional testing into more distinct “benign” and “pathogenic” categories provides clinicians with the necessary direction for next steps. Besides the above-mentioned genetic analyses, copy number variations (CNVs), which can contribute to the disease phenotype, can be assessed by chromosome microarrays. These, too, come in different flavors, and the clinician has to know which microarray platform is most suitable to the clinical context.38 There are a number of other factors that can modulate the phenotype–genotype correlation, assuming there is one, including mosaicism,39,40 epigenetic factors, digenic defects, and epistatic modulation (Abraham and Butte, JACI In Practice, 2020, In Press).
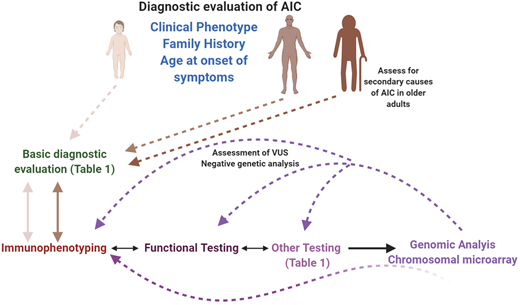
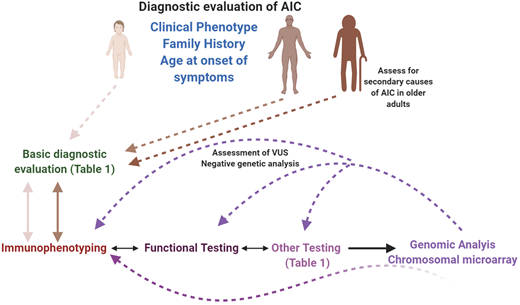
Diagnostic evaluation of AICs in pediatric and adult patients. A systematic, interdisciplinary approach can facilitate a timely and accurate diagnosis in these patients. All patients should receive a basic evaluation (expanded in Table 1), followed by immunological assessment (immunophenotyping, functional, and other specific assays), based on clinical phenotype, family history, and age of onset of symptoms. Molecular/genetic testing can be pursued either in parallel to the immunological assessment, depending on the clinical phenotype, or sequential to it. If a molecular diagnosis is not established at the first attempt, and if the likelihood of a genetic defect is high, recommend an iterative approach, which can also be used when characterizing VUSs. In the older adult, secondary causes of AIC should be eliminated before considering an intrinsic immune anomaly as the cause of the phenotype. This figure was created with BioRender.com.
Diagnostic evaluation of AICs in pediatric and adult patients. A systematic, interdisciplinary approach can facilitate a timely and accurate diagnosis in these patients. All patients should receive a basic evaluation (expanded in Table 1), followed by immunological assessment (immunophenotyping, functional, and other specific assays), based on clinical phenotype, family history, and age of onset of symptoms. Molecular/genetic testing can be pursued either in parallel to the immunological assessment, depending on the clinical phenotype, or sequential to it. If a molecular diagnosis is not established at the first attempt, and if the likelihood of a genetic defect is high, recommend an iterative approach, which can also be used when characterizing VUSs. In the older adult, secondary causes of AIC should be eliminated before considering an intrinsic immune anomaly as the cause of the phenotype. This figure was created with BioRender.com.
An observational clinical study to identify monogenic defects in pediatric Evans syndrome is currently open (NCT03912129), and such studies will continue to add to the body of knowledge on the genetics of AICs. A recent study of 18 children with Evans syndrome who did not have FAS-associated ALPS (FAS) revealed that close to 38% had single gene defects, including CTLA4 and LRBA defects. In the same study, of 48 children, 30 (62%) had FAS-associated ALPS, another IEI.41
AICs in IEIs
Although the infection phenotype was the first association made with IEI, over the last several years, numerous single-gene defects with immune dysregulation have been described and well documented.42-44 The spectrum of immunological dysregulation can range from organ-specific autoimmunity to AICs, lymphoproliferation, and susceptibility to neoplastic disease. In many of the primary immune regulatory disorders, a multiplicity of these phenotypes are present or develop over time. The range of cytopenias can vary, and knowing the molecular defect can facilitate targeted and timely treatment. The pathophysiology behind the cytopenias, affecting one or more arms of the hematopoietic compartment, is multifaceted, and here again, the molecular defect and immunological phenotype may be helpful in determining management and therapy. AIC can also develop secondary to potentially curative therapy, such as HCT for IEI, as a result of conditioning regimens, incomplete immune reconstitution, and/or viral infections, and it requires similar approaches for management.45 Single-lineage AIC includes only AIN, AIHA, or ITP when presenting independently, or multilineage disease, such as Evans syndrome, may be present. AIHA could be associated with either warm or cold autoantibodies. Similarly, for AIN and/or ITP, autoantibodies may be detected, which may help facilitate treatment decisions.
Returning to the clinical case
CTLA-4 haploinsufficiency was initially not included in the differential diagnosis for this patient, because the genetic defect had not been identified or described when the patient first presented. However, with hindsight, the phenotype fits the genotype. The laboratory evaluation as described above would have been useful to further narrow the diagnostic possibilities, had they been easily available at the time. The lack of effective CTLA-4–based regulation of the immune response results in aberrant activation of effector T cells, ineffective and dysregulated Treg function, and lymphocytic infiltration of organs. Patients with CTLA-4 haploinsufficiency have been shown to have a progressive loss of B cells with accumulation of these in various nonlymphoid organs. There is also an expansion of CD21dim B cells, which has previously been reported to be associated with autoreactivity and chronic graft-versus-host disease.46,47 Penetrance of CTLA4 variants is incomplete and is thought to be seen in two-thirds of cases.11 Also, the phenotype can vary depending on whether only the CTLA4 gene is involved or there is a larger deletion involving CTLA4 and other genes in proximity.12
Although this patient received several “nonspecific” immunomodulatory therapies, including natalizumab for an incorrect diagnosis of multiple sclerosis, rituximab, steroids, intravenous immunoglobulin, and the identification of a molecular diagnosis allowed institution of a specific and personalized treatment. This case is very similar to one reported in the literature of a patient with the same pathogenic variant in CTLA4 who had many similarities in clinical phenotype and was treated with daclizumab, targeting CD25, which resulted in further suppression of Treg function and worsening of symptoms.48 The published case highlights the pitfalls of randomly instituting immunotherapy without fully understanding the mechanism of pathogenesis of disease. Abatacept is a fusion drug of the extracellular domain of CTLA-4 with the Fc portion of an immunoglobulin; it is available and approved for the treatment of rheumatoid arthritis, and it acts as a pharmacologic replacement for the ineffective CTLA-4 in these patients and is also efficacious in LRBA deficiency.49,50 Sirolimus, a mammalian target of rapamycin inhibitor, can also potentiate Treg function by expanding Tregs and maintaining their immunosuppressive function, and it has been used with some success to mitigate the autoimmune manifestations in some patients. Other drugs that may be effective in CHAI include chloroquine or hydroxychloroquine, a lysosomal inhibitor (because CTLA-4 is cycled through the endolysosomal compartment before being expressed on the cell surface), though its effect in this disease has not been studied.6 HCT has also been employed in a few select patients with CHAI with overall success, though there have been adverse outcomes in a few patients and reports of graft-versus-host disease in others.6,51
Approach to diagnostic evaluation of AICs in pediatric and adult patients
IEIs, though representing germline genetic defects of the immune system, can manifest at any age, ranging from infancy to adulthood. However, the likelihood of an underlying genetic defect is higher in infants and young children with AICs than in adults in the fourth decade of life and beyond, which is not to categorically state that IEIs cannot manifest in adults. In adults, statistically speaking, monogenic defects are more likely in younger adults than in older individuals. The case presented here provides an example of a patient who was diagnosed with CTLA-4 haploinsufficiency in early adulthood.
Therefore, the diagnostic evaluation of AIC in children and young adults should involve a detailed family history, immunological evaluation (as described above and in Table 1 and Figure 2), and early genetic testing. On the basis of this information, treatment can be tailored to treat the molecular defect, if identified. If a genetic diagnosis is not readily obtained, an iterative diagnostic approach, as described above (Figure 2), should be employed, especially for refractory or severe AIC.
In older adults, the cause of AIC is more often than not likely to be secondary; however, a thorough evaluation should include basic immunological testing (Table 1 and Figure 2) before initiating significant immunosuppressive therapy (baseline). If there is a family history, genetic testing is recommended along with further evaluation of other affected family members, especially younger individuals. It is also helpful to obtain information on other coexisting conditions and treatments that could potentially account for the AIC, especially in patients with no family history. The level of advanced immunological testing in such older individuals is dependent on the age of onset, clinical and family history, severity of disease, and whether the AIC can be correlated with other conditions or treatments.
Use of targeted immunotherapies in IEIs and lessons learned from CTLA-4 haploinsufficiency for checkpoint inhibitor immunotherapy in cancer
A significant advantage of improved genetic testing for IEIs has been the repurposing of targeted immunological therapies for management and treatment of these diseases.52,53 They have added substantially to the therapeutic armamentarium, which had previously for decades consisted of antibiotics, steroids, generic immunosuppressants, immunoglobulin therapy, and HCT. Because curative gene therapy is available for only a small subset of IEIs,54 it has been more urgent to expand the therapeutic options for patients with various clinical phenotypes, including immune dysregulation. These have been added to the existing standard immunomodulatory and immunosuppressive agents (Table 2).
Abatacept, as previously described, has been effective in many CTLA-4– and LRBA-deficient patients50 and represents a new frontier in personalized medicine in these disorders. It has been used off-label for the last few years for various autoimmune and inflammatory manifestations of these diseases, though there is currently a phase 1/2 randomized, double-blind, placebo-controlled clinical trial open for abatacept in treatment of the chronic cytopenias in CTLA-4 haploinsufficiency (NCT03733067).
The patients with IEIs have provided many valuable lessons on the relevance of molecular pathways in the immune system and the consequences of targeting these in other diseases, such as cancer. In this article, the significance of blocking CTLA-4 is of particular relevance.55-57 Ipilimumab is a therapeutic monoclonal antibody directed against CTLA-458 that has been approved for the treatment of malignant melanoma, particularly metastatic disease. Although this treatment has had dramatic effects on the survival of patients with this disease, a sizable proportion have developed immune-related adverse events,59,60 which, though not dissimilar from those experienced by patients with CHAI, including colitis and lymphocytic infiltration, is sufficiently different in its manifestations. These adverse events require management with other immunomodulators. Therefore, pharmacological/biological manipulation of the immune system would benefit from the study of the IEIs, which offers a window into the consequences of immune manipulation and could facilitate the rational design of newer therapeutic interventions.61,62
Summary
Parallel to the discovery of new monogenic IEIs, including those associated with immune dysregulation, there has been a growth in the number of immunological laboratory assays available to phenotypically and functionally characterize various immune cell subsets and their alterations, which can be used to facilitate correlations of both genotype and phenotype. These immune assessment assays, coupled with genetic testing, have supported the diagnosis of the molecular basis of AICs in children and adults. The case described in this article highlights the importance of the intersection of hematology with immunology and the need for dialogue between these specialties in the care of patients with complex immunological diseases.
Correspondence
Roshini S. Abraham, Department of Pathology and Laboratory Medicine, Nationwide Children’s Hospital, 700 Children’s Dr, Columbus, OH 43205; e-mail: roshini.abraham@nationwidechildrens.org.