

Abstract
With recent advances in genetic sequencing and its widespread adoption for clinical diagnostics, the identification of a primary immunodeficiency (PID) as the underlying cause of diseases presenting to hematologists including refractory autoimmunity, cytopenias, immune dysregulation, and hematologic malignancy, is increasing, particularly in the adult population. Where the pathogenic genetic variants are restricted to the hematopoietic system, selected patients may benefit from allogeneic hematopoietic stem cell transplantation (allo-HSCT). Although it is generally accepted that early allo-HSCT (ie, in infancy or childhood) for PID is preferable, this is not always possible. The clinical phenotype of non–severe combined immune deficiency forms of PID can be very heterogeneous, in part because of the high number of genetic and functional defects affecting T, B, and natural killer cells, neutrophils, and/or antigen presentation. As a result, some patients have less severe disease manifestations in childhood and/or a later de novo presentation. For others, a delayed diagnosis, lack of a genetic diagnosis, or a previous lack of a suitable donor has precluded prior allo-HSCT. Specific issues which make transplantation for adult PID patients particularly challenging are discussed, including understanding the natural history of rare diseases and predicting outcome with conservative management alone; indications for and optimal timing of transplant; donor selection; conditioning regimens; and PID-specific transplant management. The role of gene therapy approaches as an alternative to allo-HSCT in high-risk monogenic PID is also discussed.
Learning Objectives
Understand and evaluate the role of allo-HSCT in adults with PID; although allo-HSCT can be curative, patient selection and optimal timing of transplant is complex
Understand the importance of always taking a careful family history in younger adults presenting with refractory autoimmunity, HLH, or lymphoproliferation/lymphoma to help identify an underlying PID
Understand the need to integrate genetic, laboratory, clinical, and family history data to predict prognosis
Understand that alternative therapies instead of, or as a bridge to, transplant are increasingly available
Clinical cases
Figure 1 illustrates the clinical course of 2 adult primary immunodeficiency (PID) patients with (i) autoimmune lymphoproliferative syndrome and (ii) hypomorphic Rag2 deficiency (combined immune deficiency).1 Both patients presented in childhood or adolescence with anemia (autoimmune hemolytic anemia in patient 1 and red cell aplasia in patient 2). Both subsequently underwent allogeneic hematopoietic stem cell transplantation (allo-HSCT) in adulthood.
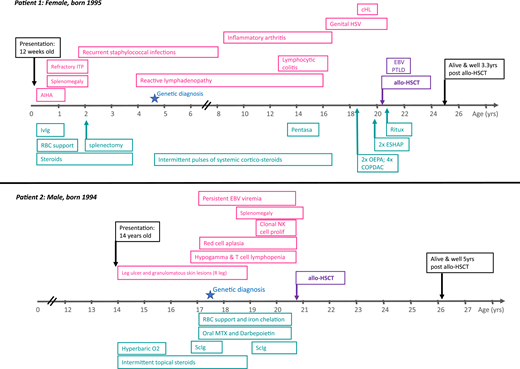
Patient 1 presented with autoimmune hemolytic anemia at the age of 12 weeks, which was successfully treated with steroids and intravenous immunoglobulin (ivIg). However, refractory immune thrombocytopenic purpura (ITP) and splenomegaly developed soon after requiring splenectomy at the age of 2 years. There was a known family history of autoimmune lymphoproliferative syndrome affecting her mother and 2 brothers. Genetic diagnosis was made in early childhood. Childhood and adolescence were complicated by inflammatory arthropathy, colitis secondary to lymphocytic infiltration of the gut mucosa, extensive genital herpes simplex virus infection, and recurrent, widespread reactive lymphadenopathy. Classical Hodgkin lymphoma (HL) was diagnosed on inguinal lymph node biopsy when the patient was 18 years old. The decision to proceed to allo-HSCT was made after relapse of HL 2 years later at the age of 20 years.
Patient 2 initially presented at 14 years of age to dermatology with a nontraumatic leg ulcer and localized granulomatous skin lesions. There was no response to hyperbaric oxygen or topical steroids. Other therapies were declined. There was no family history of immunodeficiency. At 16 years, he was noted to have hypogammaglobinemia and T-cell lymphopenia. He was started on immunoglobulin replacement therapy but soon after developed severe, refractory red cell aplasia. Two years later, he developed a clonal natural killer (NK)-cell proliferation (no T-cell clone detected), and genetic diagnosis was confirmed. He was then referred for allo-HSCT, which was performed at the age of 20 for NK-cell clonal proliferation and associated bone marrow failure.
Introduction
For the last 30 years, allo-HSCT has been considered the standard of care and the major therapeutic option for children with serious inborn errors of immunity or PID.2 Early transplant is particularly important for infants or children presenting with serious or life-threatening infections, because without definitive treatment, patients with severe PID, such as severe combined immune deficiency (SCID), rarely survive beyond 1 year of age. Until recently, the vast majority of transplant procedures for PID were performed in childhood, with clinical expertise in these rare diseases residing almost exclusively in pediatric specialist centers.
However, as an increasing number of nontransplanted PID (almost all non-SCID) patients are now surviving, or only being diagnosed, beyond childhood, most patients with PID are now found within the adult population.3 The role of allo-HSCT for adults is therefore being carefully evaluated.
Uncorrected PID can lead to recurrent, progressive, or life-threatening infections, autoimmunity, autoinflammatory manifestations, and malignant disease (typically lymphoma or epithelial neoplasia). This places a significant burden on health care resources with frequent hospital admissions, the need for ongoing antimicrobial therapy, and expensive therapy including biological-modifying drugs (eg, monoclonal antibodies) and immunoglobulin replacement therapy. The result can be poor quality of life and early death.
Within the last 3 years, evidence has been published that demonstrates selected adults with PID can be transplanted safely using reduced intensity conditioning regimens, achieving outcomes equivalent to that achieved in pediatric transplant practice.4,5 However, questions remain regarding the indications for and optimal timing of transplant in adults.
Basic principles of allo-HSCT for nonmalignant disease
Allo-HSCT allows the replacement of defective or dysregulated recipient immune and hematopoietic cells with long-term repopulating cells from a healthy donor. Apart from gene therapy, in the case of selected monogenic forms of PID, it is the only potentially curative therapy. PID patients undergoing allo-HSCT (as for other nonmalignant diseases) do not benefit from the graft-versus-malignancy effect unless they have a prior history of blood cancer (most commonly lymphoma). The prevention of graft-versus-host disease (GVHD) and successful long-term immune-hematopoietic engraftment are therefore of paramount importance. Long-term stable donor chimerism in the previously affected cell lineage/s and correction of the clinical phenotype is the ultimate goal of transplant.
Major factors influencing outcome after allo-HSCT are broadly independent of the underlying disease and include preceding comorbidity, active infection at the time of transplant, end organ function, type of donor, and age of the patient.6,7 For PID patients, the shorter the time from onset of clinical symptoms to transplant, the lower the risk of developing resistant or refractory infections (bacterial, viral, or fungal) and end organ damage caused by uncontrolled inflammation or autoimmunity.
Who and when?
One of the biggest challenges for hematologists and immunologists looking after adults with PID is knowing which patient and when to refer for consideration of allo-HSCT. The clinical decision is straightforward if the underlying condition is known to be life-threatening or life-limiting, and the patient has a predicted poor prognosis with conservative management alone. For rarer forms of PID, the natural history with conservative management alone is often not known, which necessitates careful, shared decision making with a wider team of specialist health care professionals, the patient, and their family. This is essential for the patient to be fully informed of the risks and uncertainties. However, as with all potential allo-HSCT recipients, for maximal benefit, transplant must occur before serious end organ damage has occurred, which could make the risk of transplant unacceptably high. With respect to comorbidities, the HCT-CI score (hematopoietic stem cell transplantation comorbidity index, a validated comorbidity index predicting high risk patients for HSCT in the setting of hematologic malignancies) has recently been validated as having predictive value for patients with nonmalignant diseases including PID,8 with the caveat that most patients included in the analysis were children.
Background on natural history of PID in adults and data in allo-HSCT
Every year an international expert committee provides a genotypic and phenotypic classification of all human inborn errors of immunity. There are now 406 distinct clinical disorders with 430 different gene defects,1 and although individually rare, these disorders are enriched in patients with hematologic malignancies and autoimmune cytopenias. In the more recent classifications, the concept of pure immunodeficiencies with predisposition to infections has been replaced with newly described autoimmune, autoinflammatory conditions or syndromal disorders associated with immunodeficiency. Many of these disorders can be cured by allo-HSCT, whereas in syndromal disorders, only the hematopoietic portion of the disease can be corrected, which may nevertheless be indicated with improved survival and quality of life in selected patients.
PID patients who survive to adulthood without having undergone an allo-HSCT in infancy or early childhood will almost all have some residual immune function, even if grossly abnormal. Therefore, most nontransplanted adult PID patients have non-SCID PID, such as combined immune deficiency (CID, affecting both cellular and humoral immunity), CID with syndromic features, predominantly antibody deficiencies, diseases of immune dysregulation, phagocytic/innate immunity disorders, and autoinflammatory disorders.1 In many of these, the predominant clinical features include immune dysregulation, such as refractory autoimmunity, lymphoproliferation, or autoinflammation; the presentation may be much later, and the role and timing of allo-HSCT are currently less clear.
Studies that address the long-term prognosis of PID patients are very difficult to perform, but natural disease outcome data are desperately needed to be able to determine the optimal role of allo-HSCT,9 particularly with newly described or PID with a wide phenotypic heterogeneity in disease severity. For example, most patients with common variable immunodeficiency (CVID), the most common form of PID in adults, have an excellent prognosis and few complications if receiving adequate immunoglobulin replacement therapy, whereas others with additional features of immune dysregulation develop severe complications of the lung and liver and have a very poor prognosis.10 Published retrospective data for HSCT in adult patients with complex CVID have indicated worse outcomes than for other PID patients after transplant,11 underlining the need for better information to select patients and timing for HSCT in CVID. For other PID subtypes, there is accumulating evidence of continued disease progression throughout adulthood, including chronic granulomatous disease (CGD), X-linked lymphoproliferative syndrome, and CD40 ligand (CD40L) deficiency,12-15 resulting in serious morbidity and mortality in these studies. Importantly, for other PID, such as dedicator of cytokinesis 8 (DOCK8) deficiency, recent studies have indicated that a plateau in overall survival may be reached in adulthood, but nevertheless, there is a progressive fall in event-free survival with conservative management alone.16
There is a growing body of publications detailing outcome data on approximately 200 cases of allo-HSCT for PID in adults.4,5,11,17-25 Additional data on a similar number of adults have been published in abstract form. In most published series, the overall survival was equivalent to that achieved in children and infants being in excess of 80% at a median follow-up of between 14 months and 5 years. Most data relate to patients with CGD, hemophagocytic lymphohistocytosis (HLH), and GATA2 deficiency, for whom the data are therefore most robust. The published data are summarized in Table 1.
Summary of published allo-HSCT outcome data for adult (and adolescent) PID patients
| Reference | No. of patients | PID subtype | Age at HSCT, y (range) | Donor (no.) | Conditioning | OS | EFS | TRM | Engraftment | Median follow-up (range) |
|---|---|---|---|---|---|---|---|---|---|---|
| Albert et al 20184 | 18 | 6 CGD 12 other PID | 18 y (15-22) | MRD (2) MUD (5) 1Ag MMUD (11) | Full Bu/Cy (2); Full Bu/Flu (1); Sub Bu/Flu (7); Flu/Mel ±TT (1); Treo/Flu ±TT (7). All had serotherapy. | 94% | 94% | 6% | 100% | 5 y (2-9 y) |
| Fox et al 20185 | 29 | 11 CGD 18 other PID | 24 y (17-50) | MRD (11) MUD (13) 1Ag MMUD (5) | Flu/Mel/Alem (20) Flu/Bu/ATG (8) Flu/Bu/Alem (1) | 89% at 1yr 85% at 3yrs | 90% | 14% | 100% | 3.5 y (4 mo to 12 y) |
| Jin et al 201818 | 8 | Primary HLH | 25 y (18-54)* | HaploID (6) MUD (2) | TBI/VP16/Cyclo (6); VP16/Flu/Bu/ATG (2) | 88% | NS | 12% | 100% | 27 mo |
| Leiding et al 201819 | 5 (AYA) 10 (ped <12 y) | STAT1 GOF | 29 y (18-35) 8 y (1-17) | MRD (4); MUD (8); MMUD (1); HaploID (2)‡; UCB (2, 2‡). | Flu/Mel/Alem (4); Bu/Cy (3); Flu/Bu or Treo/ATG or Alem (6); Various other (4‡) | 20% at 1yr (AYA) 60% at 1yr (ped) 40% at 1yr (all) | NS NS NS | NS | 50%† | NS |
| Parta et al 201720 | 17 (AYA) 20 (ped) | CGD | 24 y (18-32) 8 y (4-17) | MRD (6); MUD (30); MMUD (1) | Bu/Alem (6); Bu/TBI/Alem (31) | 82.5% (all) | 80% (all) | 17.5% (all) | 85% (all) | 3.4 y (range, NS) |
| Shah et al 201721 | 7 | DOCK8 Defic | 20 y (7-25) | HaploID (7) | Flu/Bu/Cy + low dose TBI (7) | 86% (all) | NS | 14% | 100% | 2 y (3 mo to 5.7 y) |
| Fu et al 201622 | 30 | 1° HLH; EBV- HLH; Tu-HLH; Undef- HLH | 23 y (14–52) 19 y (14–55) 24 y (14–44) 29 y (16–32) | HaploID (23); MRD (6); MUD (1). | Bu/Cyclo/VP16 (6) for MRD TBI/Cyclo/VP16 + ATG (24) for HaploID/MUD | 63.3% at 2 y (100% in 1° HLH; 64% in EBV-HLH; 17.7% in Tu-HLH; 25% in Undef) | NS | 100% | 26 mo | |
| Wehr et al 201511 | 14 (adult) 11 (ped) | CVID | 34 y (18-50) 14 y (8-17) | MRD (14); MUD (10); MMUD (1) | BCNU/Flu/Mel (5); Flu/Mel (7); Flu/Mel/Treo (2); Flu/Bu ±TT (4); Bu/Cy ±AraC (6); Flu/Cy (1). | 57% (adults) 52% (all patients) | 44% at 1 y (all) | 79% (adult) | NS | |
| Grossman et al 201423 | 14 | GATA2 Defic | 33 y (15-46) | MRD (4); MUD (4); UCB (4); HaploID (2). | Flu/low dose TBI (8); Flu/Cy/low dose TBI (6). | 57% (all) | NS | 28% | 100% | 3.5 y (1-5 y) |
| Gungor et al 201424 | 13 (adult) 43 (ped) | CGD CGD | 21 y (18-39) 9 y (0.8 – 17) | MRD (21); MUD (25); MMUD (10). | Flu/Bu ATG or Alem (all). | 92% (adult) 96% (all) | 91% (all) | 7% | 93% (adult) | 21 mo |
| Spinner et al 201425 | 21 | GATA2 Defic | NS (15-49 y) | NS | NS | 72% at 1 y; 65% at 2 y; 54% at 4 y | NS | NS | NS | 14 mo (0-180 mo) |
| Reference | No. of patients | PID subtype | Age at HSCT, y (range) | Donor (no.) | Conditioning | OS | EFS | TRM | Engraftment | Median follow-up (range) |
|---|---|---|---|---|---|---|---|---|---|---|
| Albert et al 20184 | 18 | 6 CGD 12 other PID | 18 y (15-22) | MRD (2) MUD (5) 1Ag MMUD (11) | Full Bu/Cy (2); Full Bu/Flu (1); Sub Bu/Flu (7); Flu/Mel ±TT (1); Treo/Flu ±TT (7). All had serotherapy. | 94% | 94% | 6% | 100% | 5 y (2-9 y) |
| Fox et al 20185 | 29 | 11 CGD 18 other PID | 24 y (17-50) | MRD (11) MUD (13) 1Ag MMUD (5) | Flu/Mel/Alem (20) Flu/Bu/ATG (8) Flu/Bu/Alem (1) | 89% at 1yr 85% at 3yrs | 90% | 14% | 100% | 3.5 y (4 mo to 12 y) |
| Jin et al 201818 | 8 | Primary HLH | 25 y (18-54)* | HaploID (6) MUD (2) | TBI/VP16/Cyclo (6); VP16/Flu/Bu/ATG (2) | 88% | NS | 12% | 100% | 27 mo |
| Leiding et al 201819 | 5 (AYA) 10 (ped <12 y) | STAT1 GOF | 29 y (18-35) 8 y (1-17) | MRD (4); MUD (8); MMUD (1); HaploID (2)‡; UCB (2, 2‡). | Flu/Mel/Alem (4); Bu/Cy (3); Flu/Bu or Treo/ATG or Alem (6); Various other (4‡) | 20% at 1yr (AYA) 60% at 1yr (ped) 40% at 1yr (all) | NS NS NS | NS | 50%† | NS |
| Parta et al 201720 | 17 (AYA) 20 (ped) | CGD | 24 y (18-32) 8 y (4-17) | MRD (6); MUD (30); MMUD (1) | Bu/Alem (6); Bu/TBI/Alem (31) | 82.5% (all) | 80% (all) | 17.5% (all) | 85% (all) | 3.4 y (range, NS) |
| Shah et al 201721 | 7 | DOCK8 Defic | 20 y (7-25) | HaploID (7) | Flu/Bu/Cy + low dose TBI (7) | 86% (all) | NS | 14% | 100% | 2 y (3 mo to 5.7 y) |
| Fu et al 201622 | 30 | 1° HLH; EBV- HLH; Tu-HLH; Undef- HLH | 23 y (14–52) 19 y (14–55) 24 y (14–44) 29 y (16–32) | HaploID (23); MRD (6); MUD (1). | Bu/Cyclo/VP16 (6) for MRD TBI/Cyclo/VP16 + ATG (24) for HaploID/MUD | 63.3% at 2 y (100% in 1° HLH; 64% in EBV-HLH; 17.7% in Tu-HLH; 25% in Undef) | NS | 100% | 26 mo | |
| Wehr et al 201511 | 14 (adult) 11 (ped) | CVID | 34 y (18-50) 14 y (8-17) | MRD (14); MUD (10); MMUD (1) | BCNU/Flu/Mel (5); Flu/Mel (7); Flu/Mel/Treo (2); Flu/Bu ±TT (4); Bu/Cy ±AraC (6); Flu/Cy (1). | 57% (adults) 52% (all patients) | 44% at 1 y (all) | 79% (adult) | NS | |
| Grossman et al 201423 | 14 | GATA2 Defic | 33 y (15-46) | MRD (4); MUD (4); UCB (4); HaploID (2). | Flu/low dose TBI (8); Flu/Cy/low dose TBI (6). | 57% (all) | NS | 28% | 100% | 3.5 y (1-5 y) |
| Gungor et al 201424 | 13 (adult) 43 (ped) | CGD CGD | 21 y (18-39) 9 y (0.8 – 17) | MRD (21); MUD (25); MMUD (10). | Flu/Bu ATG or Alem (all). | 92% (adult) 96% (all) | 91% (all) | 7% | 93% (adult) | 21 mo |
| Spinner et al 201425 | 21 | GATA2 Defic | NS (15-49 y) | NS | NS | 72% at 1 y; 65% at 2 y; 54% at 4 y | NS | NS | NS | 14 mo (0-180 mo) |
Adult ≥ 18 years at transplant. Ag, antigen; Alem, alemtuzumab; AraC, cytarabine; ATG, anti thymocyte globulin; BCNU, carmustine; Bu, busulfan; CGD, chronic granulomatous disease; CVID, common variable immunodeficiency; Cy, cyclophosphamide; def, deficiency; EBV, Epstein-Barr virus; EFS, event free survival; Flu, fludarabine; GOF, gain of function; HaploID, haploidentical; HLH, hemophagocytic lymphohistiocytosis; Mel, melphalan; MMUD, mismatched unrelated donor; MRD, matched related donor; MUD, matched unrelated donor; ns, not stated; OS, overall survival; PID, primary immunodeficiency; TBI, total body irradiation; Treo, treosulfan; TRM, transplant related mortality; TT, thiotepa; UCB, umbilical cord blood; VP16, etoposide.
Adapted from Morris and Albert41 with permission.
Age at diagnosis.
Secondary graft failure
As second or third procedure.
Because published adult transplant outcome data are limited for most PID subtypes, a large retrospective study is underway coordinated by the Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation to address this issue. Ideally, decisions about allo-HSCT would be based on prospective randomized studies. However, these are not feasible in rare and ultra-rare diseases. In an attempt to use retrospective data, a large Franco-British retrospective case-controlled study is comparing outcomes of transplanted and matched nontransplanted adults with various subtypes of severe PID with the aim of identifying whether allo-HSCT in adulthood confers genuine benefit in terms of overall survival, event-free survival, and cumulative incidence of PID-associated events. Provisional, unpublished data suggest that allo-HSCT in adult PID patients may prevent the progressive morbidity associated with PID in adults and outweigh the negative impact of transplant-related mortality (TRM) (Cheminant M, Fox T, et al, unpublished data).
Optimal timing of allo-HSCT in adults with PID
In pediatric practice, well infants/children with a diagnosis of severe PID are typically offered preemptive allo-HSCT before the development of significant infections, autoimmunity, and/or autoinflammation to reduce the risk of peritransplant complications. Where the TRM risk is small and the severity/prognosis of the underlying disease is well understood and predictable, the preemptive use of allo-HSCT is justifiable; examples of specific PID in this category include SCID, CGD, Wiskott-Aldrich syndrome (WAS), and primary HLH. For specific PID with well-described high risks of developing lymphoma or other malignancies, there may be a role for preemptive allo-HSCT after careful discussion with the patient. However, for adults with known PID who remain clinically well beyond early adulthood, the risk-benefit balance of transplant is less clear, particularly for PID subtypes with less well-studied allo-HSCT outcomes. In these circumstances, proceeding to transplant without a clear trigger or indication is not currently recommended.
Most adult PID patients referred for transplant are not at immediate risk of death but are at risk of ongoing recurrent, progressive, or life-threatening infection, autoimmunity, autoinflammation, and malignant disease, typically lymphoma or epithelial neoplasias. For other younger adults, there may have been gradual disease progression through adolescence, and therapeutic options at the time of referral include a trial of biologics and/or immunomodulatory therapies (where appropriate) or allo-HSCT. In rarer or more recently described PID, the long-term efficacy and toxicity of nontransplant treatment modalities (examples include phosphoinositide 3-kinase delta inhibitors, jak/stat inhibitors, or abatacept) is also unknown. Whether to use such agents (singly or sequentially) as a bridge to transplant by reducing PID-associated complications before transplant with the aim of reducing TRM or using them longer term, which may eventually lead to refractoriness, is open to discussion. However, some universal truths of transplantation can guide us: outcomes are generally better when disease is in remission, and the recipient is younger and has less comorbidities at the time of transplant. Such uncertainty requires hematologists and immunologists to work together effectively to build consensus and where possible recruit patients to international studies.
It is well understood that patients proceeding to transplant incur an immediate risk associated with transplant conditioning and immunosuppression. If the TRM is high because of comorbidities at the time of transplant, the potential benefit from transplant can be lost for a given individual. Because many patients with PID have several comorbidities at the time of transplant, early differences in survival between transplanted and nontransplanted patients can be small or nonexistent because of the negative impact of a high TRM.
There are clear data demonstrating that within pediatric cohorts, transplanting earlier in the disease course improves outcomes.26-29 However, in adolescent and young adult cohorts, the impact of age appears less significant.4,5 There are very few older adults (>50 years) included in published series detailing outcome after allo-HSCT for PID patients,4,5,11,18-25 and until further data are available, patients should only be transplanted with a very clear indication.
Similarly, for patients with very rare PID diagnoses or with PID for which the number of transplants performed to date are very small (eg, <10 reported cases), the data to support allo-HSCT over trials of biologics/targeted therapies is likely to be scarce.
Unfortunately, in real-world clinical practice, many adult PID patients are referred for consideration of allo-HSCT too late. Typically, referrals for transplant are triggered by a severe PID-related event (eg, a life-threatening infection, malignancy, or major organ dysfunction), which acts as a declaration of the severity of the clinical phenotype and predictor of future complications. If these complications do not respond promptly to treatment, the window of opportunity for transplant may be missed.
Indications for allo-HSCT in adults with PID
Figures 2 and 3 suggest algorithms for identifying adult PID patients who may benefit from allo-HSCT and for whom referral to a specialist transplant unit is appropriate. The process requires close multidisciplinary patient-specific discussion between the immunology and transplant teams. Additional input from pediatric immunologists who have previously cared for the patient and/or other family members may be beneficial. Decisions are made with consensus based on the experience of the group also taking account pediatric transplant data, any reported adult transplant outcomes, known natural history of the specific condition, and international advice for very rare cases.
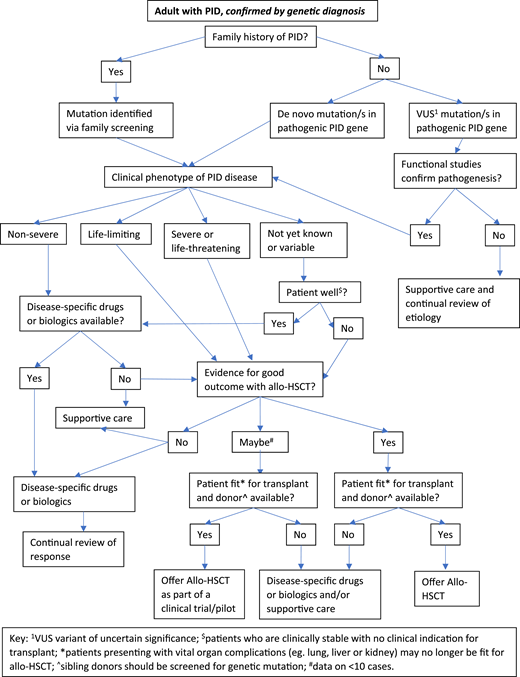
Decision flowchart (algorithm) for adult PID patients with known genetic diagnosis referred for allogeneic HSCT.
Decision flowchart (algorithm) for adult PID patients with known genetic diagnosis referred for allogeneic HSCT.
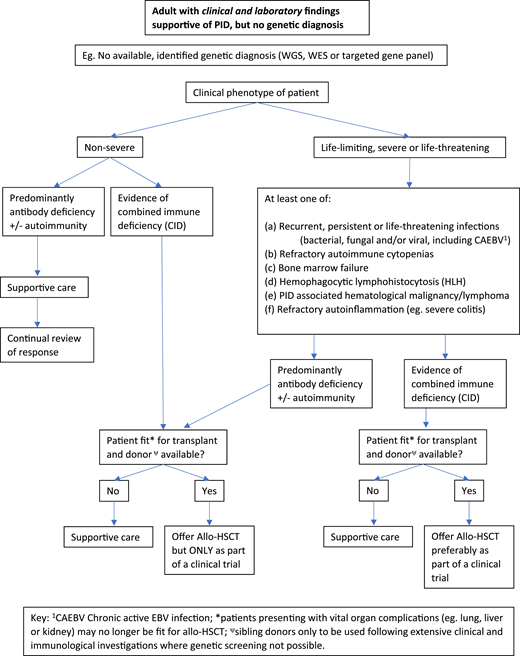
Decision flowchart (algorithm) for adult PID patients without a genetic diagnosis referred for allogeneic HSCT.
Decision flowchart (algorithm) for adult PID patients without a genetic diagnosis referred for allogeneic HSCT.
In the United Kingdom, we established a national virtual multidisciplinary team expert panel to discuss all adults with PID being considered for allo-HSCT. The multidisciplinary team serves to share expertise and discuss diagnosis, further investigations where indicated, appropriateness of transplant, conditioning regimens, and donor selection. Discussion of nontransplant treatment options is also included.
In summary, for patients who have either known pathogenic variants in PID genes (Figure 2) and/or a family history of serious PID, or clinical and laboratory findings consistent with PID (Figure 3), the currently accepted indications for transplant include the following:
Recurrent, persistent, or life-threatening infections (bacterial, fungal, and/or viral, including chronic active Epstein-Barr virus [EBV], CAEBV);
Refractory autoimmune cytopenias;
Bone marrow failure;
HLH;
PID-associated hematologic malignancy; and/or
Refractory autoinflammation (eg, severe colitis).
Evolution of clinical cases
Patient 1 was referred for allo-HSCT at the age of 20 years after relapse of HL, fulfilling the indication criteria of PID-associated hematologic malignancy/lymphoma. Patient 2 was referred for allo-HSCT after the development of a clonal NK-cell proliferation and associated bone marrow failure, fulfilling 2 of the accepted transplant indications.
Role of genetic diagnosis and family history
It is our practice to perform genetic sequencing for all PID patients being considered for HSCT if this has not already been done. Although it is sometimes necessary to proceed to HSCT for adults with PID, based on clinical disease progression alone, it is our preference to know the precise genetic diagnosis, which permits maximal prior consideration of natural history, published transplant experience, and specific extra-hematopoietic features of the disease.
For many patients, their underlying PID is well characterized, genetically, functionally, and clinically, but treatment decisions can still be challenging. As with other rare or ultra-rare diseases, predicting outcome with conservative management alone is difficult because of very small numbers of affected individuals and limited published data. Similarly, if the genetic variant has only recently been described as potentially pathogenic or is a variant of uncertain significance, therapeutic decisions need to be based on the clinical picture alone, including the cumulative rate of serious PID-related complications, family history, and quality of life.
For other adult patients, the genetic cause remains undefined despite sequencing or the urgency of transplant precludes obtaining genetic sequence results in advance. It should be remembered that serious monogenic PID are, in general, more likely to present at an early age, resulting in a lower rate of genetic diagnosis for PID presenting in adulthood.
For some PID disorders, members of the same family/kindred sharing the same genetic variants can have highly variable clinical presentations, such as nuclear factor κB1 haploinsufficiency,30 lipopolysaccharide (LPS)-responsive and beige-like anchor protein deficiency,31 and cytotoxic T lymphocyte associated protein 4 deficiency,32 so that some have severe disease and others are asymptomatic. The reasons for this are poorly understood but may relate to environmental triggers, such as infection, other genetic background, and epigenetic factors. Highly variable clinical phenotypes may also arise as a result of incomplete penetrance, variable expressivity and gain-of-function (GOF) or loss-of-function variants in the same gene, and finally, hypomorphic (leaky) defects where residual gene expression and function may be preserved. Clinical case 2 illustrates exemplifies this. Rag gene mutations can cause SCID with life-threatening presentations in infancy if recombinase activity is lacking, whereas hypomorphic mutations in the same PID gene, which permit 5% to 30% recombinase activity, result in a CID clinical phenotype, often much later in onset.33
Where possible, functional analyses of genetic diagnoses should be performed before transplant. Exceptions include rapidly deteriorating patients requiring urgent intervention and/or novel genetic variants for which no validated functional assay exists. Functional assay development for novel variants predicted to be pathogenic is also critical for the informed selection of HLA-matched family donors, who may be heterozygous carriers for the same genetic variant. This is complicated further in phenotypically variable and late-onset PID. In such cases, although TRM risks are generally higher with unrelated donors, these may be preferable.
In all cases of adult PID being considered for allo-HSCT, the final decision is a clinical one, and the absence of genetic and functional data should not necessarily preclude transplant if the clinical evidence is compelling.
Evolution of clinical cases
Both patients described in Figure 1 have heterozygous pathogenic variants in PID-associated genes. Patient 1 had a heterozygous pathogenic variant in the death domain region of TNFRSF6 gene (also known as Fas and CD95), resulting in defective apoptosis in lymphoid cells (p.I246T). This patient also had other affected family members (mother and 2 brothers) with a milder clinical phenotype, illustrating the additional complexity resulting from varied clinical phenotypes within the same kindred and/or incomplete penetrance. Patient 2 was found to have compound heterozygous-predicted pathogenic variants in the RAG2 gene [c.104G>T (Gly35Val)/het and c.814G>A (Val72Ile)/het and c.965T>C (Met 322Thr)/Het]. This patient had no family history of PID.
Donor selection
Preferred stem cell donors are 12/12 HLA-matched, cytomegalovirus (CMV) sero-matched, unaffected related donors to minimize TRM and the risk of GVHD. However, most series published to date include large numbers of matched or 1 antigen mismatched unrelated donor transplants (matched unrelated donor (MUD), typically 10/10 or mismatched unrelated donor (MMUD), typically 8/10 or 9/10), with good results. HLA-matched family members should be genetically screened if a known pathogenic variant has been identified in the recipient.
In pediatric practice, unmanipulated haploidentical transplants using PTCy (posttransplant cyclophosphamide) and αβTCR/CD19-depleted haploidentical (Haplo) transplants have achieved excellent results for infants and children with PID and other inborn errors, where no matched donors are available.34 However, there are currently insufficient published data in older PID patients to definitively support the use of haploidentical donors in preference to MMUDs. In non-PID settings, a number of prospective randomized controlled trials are being planned to determine the safety and efficacy of MMUD vs Haplo in patients >18 years at HSCT.
Conditioning regimens
PID patients surviving to adulthood typically have residual functional cellular immunity necessitating conditioning to permit engraftment of allogeneic stem cells and prevent graft rejection; as a consequence, unconditioned transplants are not indicated.
To limit TRM in older PID patients, either reduced toxicity or reduced intensity conditioning is recommended in all cases. However, it is increasingly evident that achieving stable donor chimerism is important for PID transplants, and this is not always reliably achieved with reduced intensity conditioning regimens. Typically, 10% to 20% of children undergoing allo-HSCT for PID require a second procedure or donor lymphocyte infusion (DLI), with HLA mismatch and the use of reduced intensity conditioning being significant risks.35
Most experience to date has been with fludarabine/busulfan (Flu/Bu), fludarabine/melphalan (Flu/Mel), or fludarabine/treosulfan (Flu/Treo)-based reduced intensity conditioning incorporating serotherapy (alemtuzumab or ATG) for in vivo T-cell depletion. Recent improvements in conditioning regimens for nonmalignant diseases such as PID have primarily come from pediatric centers and include the introduction of treosulfan,36,37 the use of targeted busulfan,24,38 and personalized serotherapy dosing.34,35,39
The use of PTCy or αβTCR depletion to remove alloreactive T cells has facilitated the use of haploidentical donors in older recipients with hematologic malignancies, without prohibitive risks of GVHD and/or graft rejection.40 Similarly, in pediatric series, these approaches have been used with excellent results in nonmalignant disease and PID.41-43 However, as the cumulative experience transplanting older adults with PID using haploidentical donors remains very small, further studies are warranted.
Evolution of clinical cases
Patient 1 had an unaffected sibling who was HLA matched and CMV matched (+/+). Patient 2 had a fully matched unrelated donor, also CMV matched (−/−). Both patients received reduced intensity conditioning with fludarabine, melphalan, and alemtuzumab, together with single-agent cyclosporine as GVHD prophylaxis.
Adult PID-specific transplant management
Additional pretransplant investigations are indicated in many PID patients to document the presence or absence of specific pathogens, antimicrobial sensitivities, and/or degree of organ specific inflammation. Examples include the following: colonoscopy and gastrointestinal tract imaging for patients with inflammatory colitis or prior chronic norovirus or cryptosporidial diarrhea; high-resolution computed tomography chest imaging, bronchoscopy, lavage, and/or biopsy in patients with granulomatous lymphocytic inflammatory lung disease, previous aspergillosis, or atypical mycobacterial infections; and liver biopsy, fibroscan, and/or measurement of portal pressures in patients with abnormal liver function tests and a history of nodular regenerative hyperplasia or autoimmune hepatitis. Other pretransplant investigations occasionally indicated are lumbar puncture with cerebrospinal fluid culture and biopsies of atypical granulomatous lesions, where relevant, to exclude or identify persistent pathogens requiring tailored antimicrobial prophylaxis throughout the transplant period.
Where possible, control of autoimmunity or inflammation should be achieved before transplant. This reduces the inflammatory cytokine milieu at the time of transfer of allogeneic cells, thus reducing the risk of acute GVHD and ensures end organ function is optimized before transplant. Preexisting PID-associated malignancies should be treated and in remission as per routine practice in HSCT for lymphoid malignancies. For patients with EBV handling disorders, the inclusion of rituximab in the conditioning regimen can bridge the gap until functional immune reconstitution is achieved after transplant.
Specialist infectious disease advice should be sought for patients with a history of refractory or atypical infections preceding transplant. Regular posttransplant monitoring for recurrence of previous, persistent, or latent opportunistic pathogens is indicated on a per-patient basis. We recommend the continued use of prophylactic antimicrobials until cessation of immune suppression as GVHD prophylaxis or treatment.
Splenomegaly in PID is not uncommon and has multiple causes. A number of studies have clearly demonstrated that poor graft function and hematologic recovery after allo-HSCT is associated with splenomegaly.44,45 Although prior splenectomy may improve hematologic recovery after transplant, it is associated with perioperative complications and lifelong impaired immunity to encapsulated bacteria; therefore, it is only rarely performed.
Recurrence or de novo autoimmunity after transplant has also been reported in PID transplant cohorts, particularly in the context of mixed chimerism,6,46 which is discussed below. The exact pathophysiology underlying persistent or late onset autoimmunity is poorly understood.
Evolution of clinical cases
Patient 1 developed early EBV-associated post transplant lymphoproliferative disease at 2 months after transplant. She responded to 4 cycles of rituximab and prompt withdrawal of immune suppression, without the development of GVHD. EBV viremia (without lymphadenopathy) resolved by 5 months after transplant.
Importance of donor chimerism after transplant
Conditioning regimens using reduced doses of cytoreductive agents are better tolerated in older recipients with higher comorbidities, but they carry the risk of failing to eradicate host hemopoiesis. This can result in mixed chimerism in differentiated lineages including neutrophils, B cells, red cells, and platelets, although the T-cell compartment is most commonly affected. Mixed T-cell chimerism after transplant can follow re-expansion of residual recipient T cells not effectively depleted by lymphodepleting conditioning +/− serotherapy (alemtuzumab or ATG).47,48
For very long-term functional immune reconstitution, multilineage stable donor chimerism is considered optimal, although full donor chimerism in any given lineage is not per se required for correction of a functional deficit. However, mixed donor/recipient stem cell chimerism is expected to only partially correct the underlying immunologic disorder with a risk of late complications or disease recurrence.
During the first year after transplant, chimerism may fluctuate. The use of DLI to promote/force conversion from mixed chimerism to full donor chimerism should be evaluated carefully because it carries with it a risk of GVHD. Previously the use of DLI was only considered where worsening mixed chimerism raised concerns of incipient graft rejection. However, recent data suggest that persistent mixed chimerism after transplant is associated with poorer event-free survival as long as 20 years after transplant (E.C.M., unpublished data, October 2020) and late complications such as autoimmunity.6,46 Future studies should investigate the role of prophylactic or preemptive DLI early after transplant in the setting of allo-HSCT for PID.
For patients with neutrophil defects, achieving high level stable myeloid chimerism is essential. Studies in male patients and female carriers of the X-linked form of CGD have demonstrated that symptoms arising from inflammation or autoimmunity may be present in carriers with extreme degrees of lyonization (resulting in 20%-30% normally functioning phagocytes), although the risk of serious infection is rarely present if >10% of circulating neutrophils are functional.49,50
Where at all possible, known carriers should not be used as stem cell donors in PID, because even with full donor chimerism, full immunologic correction may not be achieved.
For rarer non-SCID PID and immune dysregulatory syndromes such as activated PI3k Delta syndrome, CTLA4 deficiency, LRBA deficiency, and STAT1 GOF, there are insufficient data regarding the optimal chimerism required for long-term immunologic correction and cure, although insight into the disease-specific pathophysiology may predict which lineages are critical to correct.19,51-53
How do we measure success after allo-HSCT in adult PID?
In malignant disease, a successful transplant results in long-term survival, no recurrence of the original malignancy, normal hematopoietic reconstitution, no chronic GVHD, and no late complications. Recently the use of composite outcome measures such as GVHD-free, relapse-free survival or chronic GVHD-free, relapse-free survival has been included in studies of allo-HSCT.54 In parallel, the Center for International Blood and Marrow Transplant Research is developing the use of patient-reported outcomes.55 In adult PID, the indication for transplant is often the prevention of progressive decline in quality of life secondary to the accumulation of PID-related medical complications, and patient-reported outcomes are vital in evaluating success. Other important end points for PID patients include the reconstitution of normal pathogen-specific immunity, resolution of autoimmunity and/or inflammation, and reduction in future malignancy risk.
For some PID cases, functional correction of the underlying immune deficit is easy to measure, for example, the correction of neutrophil function in CGD, as measured by nitroblue-tetrazolium or dihydrorhodamine assays. In X-linked PID where carriers are asymptomatic, it is possible to achieve functional/clinical correction without full donor chimerism in the relevant lineage. In the context of gene therapy, it is relatively easy to track the gene-corrected immune cells and quantify functional correction. In allo-HSCT recipients, lineage-specific donor chimerism is a surrogate marker for functional correction, but proof of transplant efficacy relies on functional immunologic assays (eg, response to vaccination, T-cell proliferation, normalization of lymphocyte subsets) and clinical responses. For patients going into transplant with preexisting end-organ damage, it is critical that the consent process involves a clear discussion regarding which disease-associated symptoms/complications can be improved by transplant and which cannot (eg, bronchiectasis/pulmonary fibrosis, gut strictures, and extra-hematologic complications).
Evolution of clinical cases
Patient 1 is now alive and well, off immune suppression and immunoglobulin replacement therapy, with no GVHD, no recurrence of lymphoma, and employed at 40 months after transplant. Patient 2 is alive and well, off immune suppression and immunoglobulin replacement therapy, with no GVHD, normal peripheral blood counts, and at university 5 years after transplant.
Role of solid organ transplant in combination with allo-HSCT in adult PID patients with end organ failure
The incidence of end organ failure increases over time for untransplanted PID patients. As a result, a proportion of adult patients referred for transplant may already have or be at risk of imminent end organ failure. There is a small amount of published literature on the role of solid organ transplant either before or after allo-HSCT, and in selected cases, this should be considered.56 Most available data in PID is for combined liver transplant in combination with allo-HSCT. Physicians and patients should be aware that allocation of cadaveric organs for patients with uncorrected PID also requiring an allo-HSCT for which outcome data are scarce is not universally guaranteed. Because of limited availability of such organs, they are typically reserved for clinical scenarios for which the evidence of long-term benefit is strongest. Furthermore, in a subset of patients, the severity of their liver disease may not yet meet criteria for liver transplant but precludes safe allo-HSCT. This very specialist area of clinical practice would benefit from ongoing international collaboration and prospective pilot studies.
Role of gene therapy as an alternative to allo-HSCT
For patients without a matched related donor or a fully matched unrelated donor, the decision between using multiple mismatched cord units, a mismatched unrelated donor, a haploidentical donor, or to consider gene therapy (where available, eg, for X-linked SCID, adenosine deaminase deficiency, WAS, X-linked CGD, and in development for other diseases including autosomal recessive-CGD and CD40L deficiency) remains difficult, in part because of the relatively limited experience of all these modalities in older patients with PID.
The use of alternative donors has a proven safety and efficacy record in younger children with PID and in adults with hematologic malignancies. It is predicted that the use of PTCy and or αβTCR depletion in these older patients will also facilitate the safe use of allo-HSCT in a wider group of potential recipients lacking a perfect donor.
Gene therapy has been successfully used in adults for WAS57,58 and X-linked CGD59 where appropriately matched allogeneic stem cell donors were not available. Gene therapy approaches currently rely on the ex vivo modification of autologous hematopoietic stem cells with viral vectors encoding the wild-type version of the absent or mutated gene. The early clinical trials of hematopoietic stem cell gene therapy using gammaretroviral vectors resulted in clonal expansion of gene-corrected cells, mediated by potent enhancer elements in the gammaretroviral long-terminal repeats, and led to the development of leukemia in some patients.60 In addition, CpG dinucleotide promoter methylation led to silencing of transgene expression limiting durability of effective gene correction.61 The most recent trials have demonstrated much improved outcomes with the use of self-inactivating lentiviral vectors (developed to minimize the mutagenic risk) and enhanced promoter sequences.
The potential advantage of gene therapy is the requirement for less immunosuppressive conditioning and no risk of GVHD. Both of these potential benefits are important in adult PID patients, who typically have high comorbidity scores when referred for a definitive procedure. Theoretically, gene therapy approaches in adults could be preferred to allo-HSCT if long-term correction and immune reconstitution is proven to be effective and durable.
Role of allo-HSCT in adults and adolescents who have failed gene therapy
As a reflection of the profound impact of gene therapy in monogenic PIDs, including X-SCID, ADA-SCID, CGD, and WAS, some of the first ever patients who were treated with hematopoietic stem cell gene therapy have recently transitioned into adult care. The first gene therapy protocols for ADA-SCID and X-SCID used minimal or no chemotherapy conditioning because the objective was to secure T-cell reconstitution, with the expectation that T cells arising from gene-corrected stem cells would have a clear competitive advantage to successfully secure engraftment of gene-corrected cells. However, a small number of recipients of unconditioned gene therapy have failed to reconstitute humoral immunity, requiring long-term immunoglobulin replacement therapy,62 and others remain lymphopenic despite persistence of gene-corrected cells. Some of these patients, now young adults, have undergone or are being considered for either repeat gene therapy or allo-HSCT as a rescue procedure.
Understanding of the durability of gene therapy efficacy is limited by its relatively recent introduction compared with allo-HSCT for PID. As more recent gene therapy protocols have adopted targeted conditioning with improved engraftment of gene-corrected stem cells and incorporated safer and more efficient vectors, it is predicted that future results will continue to improve.
Conclusion
Recent data have established that allo-HSCT in adults PID is safe, that engraftment can be reliably achieved, and that overall survival is excellent for well-selected patients. Despite the availability of next-generation gene sequencing for a large proportion of patients, the decision to proceed to transplant remains a complex clinical decision.
There is an urgent need to inform practice further with large international multicenter studies designed to assess outcome after allo-HSCT for PID in adults, together with equivalent studies describing the natural history of these rare diseases for untransplanted patients. It is important that future prospective studies include detailed analysis of functional immune reconstitution, lineage-specific chimerism, quality of life, psychosocial impact, and late effects.
Correspondence
Emma C. Morris, Institute of Immunity and Transplantation, University College London, Rowland Hill St, London, NW3 2PF, United Kingdom; e-mail: e.morris@ucl.ac.uk.
