Abstract
The Philadelphia chromosome–negative (Ph−) myeloproliferative neoplasms (MPNs) are a heterogenous group of hematopoietic stem cell diseases characterized by activated JAK/STAT signaling and a variable propensity toward myelofibrotic and leukemic transformation. Acquisition of somatic mutations in addition to the canonical JAK2, MPL, and CALR mutations found in MPNs is an important catalyst in the clonal evolution and progression of these disorders. In recent years, our increasing understanding of the molecular landscape of Ph− MPNs has generated important prognostic information that informs our approach to risk stratification and therapeutic decision-making. This review will focus on the critical impact of genomics on our approach to management of advanced Ph− MPNs.
Learning Objectives
Recognize the acquisition of somatic mutations as a catalyst for the clonal evolution and progression of MPNs
Understand the incorporation of both clinical and molecular risk scores into the management of Ph-negative MPNs, including consideration for allo-HCT
Introduction
The classic Philadelphia chromosome–negative (Ph−) myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders typified by proliferation of myeloid lineage cell lines and include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF).1 These disorders are characterized by constitutive activation of the JAK/STAT-signaling pathway and the majority of cases will harbor canonical mutations in JAK2, CALR, or MPL.2,3 In recent years, advances in next-generation sequencing (NGS) methodologies have led to the identification of other mutations that are associated with clonal evolution and progression in Ph− MPNs. This review will focus on the practical ways in which this knowledge is informing our approach to risk stratification and therapeutic decision-making, including the timing of allogeneic stem cell transplantation.
Clinical case part 1
A 60-year-old man with PV presented with complaints of night sweats, a 10-kilogram weight loss, early satiety, and abdominal fullness over the preceding 6 months. He was still working full-time but was no longer able to exercise due to complaints of significant fatigue. His original diagnosis was made 15 years prior when he was concurrently diagnosed with a deep vein thrombosis. His disease had been controlled with hydroxyurea and he had also been on low-dose aspirin after completing a course of therapeutic anticoagulation. His physical examination revealed palpable splenomegaly 15 cm below the costophrenic angle. The complete blood count showed a hemoglobin of 11 g/dL, platelet count of 165 × 103/µL, and white blood cell count (WBC) of 15 × 103/µL (70% neutrophils, 20% lymphocytes, 3% monocytes, 3% myelocytes, 2% metamyelocytes, 1% basophils, 1% eosinophils). Peripheral blood smear was notable for leukoerythroblastosis and dacrocytes. A bone marrow biopsy was performed and the morphologic findings were consistent with progression to myelofibrosis (MF). Bone marrow blasts were not increased. Reticulin stain revealed 2+ marrow fibrosis. The bone marrow cytogenetic analysis demonstrated a normal male karyotype. NGS analysis revealed the following pathogenic variants: an ASXL1 Q910Tfs*14 mutation, an SRSF2 R94dup mutation, and a TP53 R248W mutation in addition to the previously noted JAK2 V617F mutation.
Clonal evolution of MPNs and prognostic impact of comutations
Myelofibrotic transformation is recognized as an advanced presentation of Ph− MPNs and is associated with a negative impact on survival. MF can arise de novo (PMF) or secondary to underlying PV or ET (post-PV/ET MF), with a largely indistinguishable clinical presentation. There is significant heterogeneity in outcomes in MF with survival ranging from <2 years to more than a decade. This has spurred the development of conventional prognostic scoring models over time, including the International Prognostic Scoring System (IPSS) and the Dynamic IPSS (DIPSS), which are largely derived from clinical variables. The DIPSS-plus model also incorporates clonal cytogenetic abnormalities (Table 1).4-6
Over the last decade and a half, considerable progress has been made in identifying the genotypic drivers that facilitate clonal expansion and clonal dominance in the Ph− MPNs, and characterizing their intersection with aging, clonal hematopoiesis, epigenetic events, host genetic, and environmental factors in modulating the MPN phenotype and impacting prognosis.7-9 For example, early on, there was a particular focus on the role of JAK2V617F in disease progression and a positive correlation was established between the JAK2 allelic burden and progression from PV to MF.10 More recent studies have demonstrated that beyond the canonical mutations in JAK2, MPL, and CALR, mutations in myeloid malignancy driver genes, including those involved in DNA methylation (IDH1/2, TET2, DNMT3A), chromatin modification (ASXL1, EZH2), RNA splicing (U2AF1, SF3B1, SRSF2), and DNA repair (TP53, PPMID) are implicated in the pathogenesis of Ph− MPNs. In general, patients with a sole canonical MPN mutation were at low risk of disease progression; additional somatic mutations, however, were associated with a higher risk of an MF phenotype and/or transformation to acute myeloid leukemia (AML) and inferior overall survival (OS).11 In PMF, the presence of mutations in ASXL1, EZH2, SRSF2, U2AF1, and IDH1/2 portends a high risk for shortened OS or leukemia-free survival.12,13 The number of detrimental mutations was inversely correlated with OS, consistent with the aforementioned impact of multiple mutations with clonal evolution. By contrast, the presence of a mutation in exon 9 of CALR was noted to be associated with improved OS independent of other mutations and conventional risk-scoring systems.12,14
Given the critical impact that the mutational profile has on disease progression and outcomes in MF, an ongoing challenge in recent times has been development of risk-stratification models that are more comprehensive and that reflect our current understanding of the impact of genomics on prognosis and its interdigitation with clinical risk factors. Such models might provide greater nuance when considering the potential merits of pursuing potentially curative but relatively high-risk therapeutic strategies such as allogeneic hematopoietic stem cell transplantation (allo-HCT). Accordingly, newer molecularly inspired prognostic models have been developed with the goal of integrating clinical, cytogenetic, and mutational data to refine risk stratification (Table 2). The Mutation-Enhanced International Prognostic Score System (MIPSS70), MIPSS70+, MIPSS70+ 2.0, and Genetically Inspired Prognostic Scoring System (GIPSS) were developed from multicenter cohorts of PMF patients <70 years of age with a primary objective of refining risk to aid decision-making with regard to allo-HCT, but they have also been validated in adults over the age of 70 years with PMF.15-17 The Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) was specifically developed for those with post-PV/ET MF.18 The primary strength of these molecularly based models in comparison with clinically based models is the refinement in risk stratification based on knowledge of cytogenetic and molecular profile. For example, 46% of patients in the MIPSS70 high-risk category were upgraded from lower-risk IPSS categories, and 14% of those in the high-risk IPSS category were considered intermediate risk by MIPSS70.15 Ultimately, there is a critical need for prospective validation of current models that integrate clinical and genomic information with a view to identifying optimal ways to incorporate these into therapeutic decision-making in MF.
Therapeutic implications of genomics in MF
JAK inhibition
Given the aberrant activation of the JAK/STAT-signaling pathway in MPNs, JAK inhibition has become an important strategy in the management of MPNs over the past decade. Ruxolitinib, an orally bioavailable inhibitor of JAK1 and JAK2, is US Food and Drug Administration (FDA) approved for the treatment of MF, based upon data from the COMFORT-1 and COMFORT-2 trials.19,20 The agent is relatively well tolerated. Myelosuppression can, however, be dose limiting, particularly in patients with baseline cytopenias. Infectious complications requiring dose adjustment are less common and include respiratory tract infections, urinary tract infections, and herpes zoster infection. Of note, each of these infectious complications was seen at similar rates in placebo-treated patients with the exception of zoster.21 Isolated cases of serious opportunistic infections such as progressive multifocal leukoencephalopathy and viral reactivation have been reported in the literature, but whether there is a definitive link to ruxolitinib, particularly because these infections were not noted in long-term follow-up of COMFORT-1 or COMFORT-2, remains unclear.20-22
Although ruxolitinib has been shown to be very effective at reducing spleen volume and controlling the constitutional symptoms associated with MPNs, it has minimal impact on the underlying malignant clone. Mutant JAK2 variant allele frequency (VAF) was only modestly reduced in ruxolitinib-treated patients enrolled on COMFORT-1 and COMFORT-2,19,20 and the rate of progression to leukemia was not significantly changed. Longer follow-up of COMFORT-1 demonstrated that the median duration of response to ruxolitinib is 3 years, and >70% of patients are off therapy by 5 years.21 Mutational complexity negatively impacted the likelihood of response and patients with 2 or fewer mutations had a ninefold greater likelihood of response to ruxolitinib compared with patients with 3 or more mutations. In addition, patients with 3 or more mutations had a shorter time to treatment discontinuation, and poor OS.23
Currently, allo-HCT is the only treatment option with curative potential for MF. Given the potential risks associated with allo-HCT, however, an ongoing question in transplant-eligible patients is the choice of JAK inhibition as the initial treatment of choice and delaying transplantation vs proceeding as soon as feasible to an allo-HCT.
Optimal timing of allo-HCT
Given the heterogeneous clinical course seen in chronic-phase MPNs, the decision to pursue allo-HCT should be carefully considered and discussed with patients. As discussed in further detail in recent comprehensive reviews, factors to consider in addition to risk stratification of disease include age, comorbidities, and donor options.24,25 A number of large retrospective series have analyzed allo-HCT outcomes based upon clinical risk score, some of which are highlighted in Table 3.26-28 These have demonstrated that clinical risk scores such as DIPSS predict outcome even after allo-HCT. In addition, when the relative risk of death after allo-HCT for MF was compared with a cohort treated without transplant, the data suggest that the benefit of allo-HCT is most evident in intermediate-2 and high-risk patients and that low-risk patients should have transplant deferred. These findings hold up even in the JAK-inhibitor era when outcomes following allo-HCT were compared with nontransplant therapies that included JAK inhibition.28 The decision to pursue allo-HCT in intermediate-1 risk disease is less well defined and could be potentially impacted by the mutational profile.
The impact of genomics on outcomes?
Given the accumulating body of evidence that underscores the prognostic impact of specific somatic mutations in MPNs, incorporation of these data may help to identify candidates that serve to gain the most from transplant, including candidates with high-risk mutations who may otherwise have been categorized as having low/intermediate-1 disease by conventional risk-stratification models. Multiple retrospective analyses have been carried out to try and ascertain the impact of mutational status on outcome of MF patients undergoing allo-HCT. Regarding canonical MPN mutations (JAK2, CALR, and MPL), Panagiota et al found that triple-negative (those lacking mutations in JAK2, CALR, and MPL) MF patients undergoing transplant had inferior OS whereas CALR-mutated patients had improved OS.29 Of note, this may have been reflective of underlying disease biology given the favorable prognosis associated with type 1 CALR mutations even in the absence of allogeneic stem cell transplantation. More recent analyses have gone beyond evaluating the canonical MPN mutations and their prognostic impact in the allo-HCT population, to analyze the impact of additional mutations (Table 4).30-33 Kröger et al reported that having a CALR mutation was significantly associated with improved OS whereas having an IDH2 or ASXL1 mutation was significantly associated with inferior relapse-free survival (RFS).30 Stevens et al retrospectively evaluated a cohort of MF patients who underwent allo-HCT and demonstrated that the presence of 3 or more mutations in addition to a JAK2 or CALR mutation was associated with inferior outcomes regardless of DIPSS+ score. Of note, 75% of the patients in this cohort underwent a myeloablative regimen.31 In contrast, Tamari et al demonstrated that a U2AF1 mutation was associated with inferior OS and RFS whereas a DNMT3A mutation was associated with inferior RFS. Mutational complexity did not impact outcomes and MIPSS70 high-risk patients did not have inferior outcomes when compared with intermediate-risk patients. Interestingly, patients who received a myeloablative conditioning (MAC) regimen, which in this cohort was generally paired with a T-cell–depleted allograft, had a superior outcome to those who underwent a reduced-intensity conditioning (RIC) regimen.32 Ali et al reported on a cohort exclusively treated with a RIC regimen; they demonstrated that a CBL mutation was associated with inferior OS and DFS while a U2AF1 mutation was associated with increased non-relapse mortality. In addition, the high-risk MIPSS70 group had inferior OS and DFS when compared with the intermediate-risk group. Similarly, the MIPSS70+ v2.0 very high-risk group had worse OS and DFS when compared with the high-risk group.33
The conflicting data derived from these various analyses may have arisen from the relatively small sample size in the various cohorts, particularly with regard to the less common mutations, as well as the heterogeneity of conditioning regimens used. This suggests that larger cohorts of patients who are more uniformly treated need to be analyzed to more definitely answer the question of the impact of specific mutations on outcome of patients with MF undergoing allo-HCT. Ideally, as has been demonstrated in the context of other myeloid neoplasms,34,35 the optimal choice of conditioning regimen (MAC vs RIC) also deserves prospective study, especially in a fit, younger cohort of MF patients. However, these patients are significantly underrepresented in clinical practice. Given the predictive impact of measurable residual disease by molecular monitoring on relapse incidence,36 novel targeted maintenance strategies to eradicate measurable residual disease post–allo-HCT will be necessary to improve long-term outcomes in patients with MF, the majority of whom are older and will continue to be offered RIC regimens. The role of JAK inhibition as a posttransplant maintenance strategy is also unclear as there are limited data to currently guide clinicians in this setting.37
Clinical case part 2
The patient is classified as intermediate-1 risk by DIPSS/DIPSS+ and high risk by MIPSS70/MIPSS70+ 2.0. He is transitioned from hydroxyurea to ruxolitinib due to his significant splenomegaly and constitutional symptoms and is simultaneously referred for transplant evaluation given his MIPSS70 high-risk disease. He ultimately decides against pursuing allo-HCT despite being counseled about the negative implications of his high-risk molecular risk profile, both for OS and leukemia-free survival. He achieves significant clinical improvement with regard to splenomegaly and constitutional symptoms with ruxolitinib, but 18 months later, he presents to the clinic with complaints of worsening fatigue and weight loss. Physical examination is notable for worsening splenomegaly. Complete blood count is notable for a hemoglobin of 8.2 g/dL, platelet count of 96 × 103/µL, and a leukocyte count of 17 × 103/µL (51% neutrophils, 15% lymphocytes, 3% monocytes, 3% myelocytes, 2% metamyelocytes, 2% basophils, 2% eosinophils, 25% blasts). Repeat bone marrow biopsy demonstrates grade 3+ reticulin fibrosis and 26% blasts, consistent with a blast-phase (BP) MPN (MPN-BP). Cytogenetics demonstrate a monosomy 7. Repeat NGS demonstrates development of an IDH2 mutation and an additional mutation in TP53. The previously noted mutations in JAK2, ASXL1, SRSF2, and TP53 persist.
Genomics of AP and BP MPNs
The development of additional somatic mutations can drive MPNs toward leukemic transformation.11,38 The incidence of progression to BP, operationally defined as 20% or more blasts in the peripheral blood and/or marrow, varies based upon the underlying MPN. An analysis of 826 patients at the Mayo Clinic demonstrated a 20-year incidence of BP of 3.8% for ET, 6.8% for PV, and 14.2% for PMF.39 Historically long term survival of PMF patients with 5% to 9% circulating blasts, accelerated-phase (AP) MPN with 10% to 19% blasts (MPN-AP), and MPN-BP has been quite poor regardless of treatment approach including intensive induction chemotherapy or hypomethylating agent therapy.40-43 Early-phase trials evaluating hypomethylating agents in combination with ruxolitinib in patients with MPN-AP/BP have demonstrated limited impact on the natural history of disease, with median OS in the 7- to 8-month range.44,45 In addition, patients who develop MPN-BP after receiving treatment with ruxolitinib have incredibly poor outcomes. In an analysis of 589 MF patients treated with ruxolitinib, 11% of patients developed BP at median follow-up of 3 years from initiation of ruxolitinib (range, 0.1-7.6 years). Median survival was 2 months upon progression to BP. Although canonical MPN mutation status was not identified as a risk factor for progression to BP, patients with intermediate-2/high-risk disease by DIPSS or MYSEC-PM were at higher risk of progression to BP.46
The mutational spectrum of MPN-BP is distinct from that of de novo AML. Mutations in genes such as FLT3, NPM1, and CEBPA, which are typical of AML de novo, are rare in MPN-BP. By contrast, MPN-BP is enriched for mutations in epigenetic modifiers, DNA repair, and spliceosomal genes.40,47,48 The evolutionary pathway to MPN-BP is complex with evidence supporting evolution from a JAK2 V617F–mutated clone, clonal expansion of a pre-JAK2 V617F ancestral clone, or in some cases biclonal disease at the outset. The emerging information on the molecular drivers of blast transformation coupled with the increasing availability of targeted therapies has the potential to inform a molecularly based approach to treatment of these disorders.42,49 A number of therapies specific to underlying somatic mutations are either FDA approved or in clinical investigation in myeloid malignancies. For example, the IDH1 inhibitor ivosidenib and IDH2 inhibitor enasidenib are both approved for the treatment of IDH1- or IDH2-mutated AML in the first- and second-line settings, respectively. IDH1/2 mutations predict a short leukemia-free survival in MF and are significantly enriched in MPN-BP. There are emerging retrospective data to suggest that IDH-mutated patients with MPN-AP/BP can have relatively durable responses with IDH inhibitor–based treatment strategies.50 For TP53-mutated patients, hypomethylating agent–based strategies are often used especially in less fit individuals due to the very low likelihood of success from an intensive approach from a risk/benefit perspective. TP53 loss of heterozygosity is often associated with the progression to MPN-BP, and outcomes are particularly inferior in this subset even after allo-HCT.51 Preliminary reports from early-phase trials utilizing APR-246, a p53 modulator, and magrolimab, an anti-CD47 monoclonal antibody, have shown promising efficacy in TP53-mutated AML/myelodysplastic syndrome.52,53 There is a paucity of data with these novel agents in MPNs and such strategies merit investigation in TP53-mutated MPN AP/BP.
Allogeneic stem cell transplant is the only known cure for MPN AP/BP, but the overall cure rate is low. The optimal depth of remission in MPN-BP prior to proceeding with allo-HCT has not yet been defined.42 Recent reports suggest that the degree of disease control at time of allo-HCT may not have a significant prognostic impact.51,54,55 This may be due in part to the fact that deep remissions pre-allo-HCT for MPN-BP are rare, and therefore the relapse rate is uniformly high. This underscores the need for prospective novel approaches focused on this patient population.
Clinical case part 3
Given the presence of an IDH2 mutation, clinical trial options incorporating IDH2 inhibition were reviewed with the patient. He elected to participate in a clinical trial of enasidenib in combination with azacitidine and achieved a complete response of the leukemic component of his disease with histopathologic evidence of persistence of his chronic-phase MPN.56 His hematologic parameters have improved and he has now been on enasidenib-based therapy for several months. Follow-up NGS analysis demonstrated persistence of the JAK2 mutation, however, the TP53, ASXL1, SRSF2, and IDH2 mutations are no longer detectable. He has now agreed to move forward with an allo-HCT.
Summary
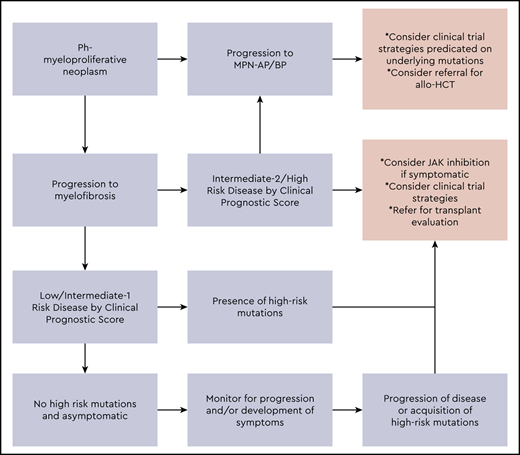
Our approach to the management of advanced MPNs is summarized in Figure 1. The integration of clinical variables and mutational data into risk-stratification efforts is paving the way for personalized prognostic modeling to identify patients who are at particularly high risk of progression and may serve to benefit the most early on, from potentially curative but relatively high-risk interventions such as allo-HCT. Many patients will be deemed ineligible for allo-HCT, and treatment of such patients remains a significant area of unmet need. A number of novel therapeutic approaches are under investigation for advanced MF, including MPN AP/BP disease (Tables 5 and 6). There is a need for ongoing investigation to define the most relevant molecular drivers of disease progression and validation of these both in non-clinical models and in early phase trials, as credible therapeutic targets (Table 7). The ultimate hope is that these efforts will significantly impact the natural history of advanced-phase MPNs.

Correspondence
Olatoyosi Odenike, Section of Hematology/Oncology, Department of Medicine, The University of Chicago Medicine, 5841 S. Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: todenike@medicine.bsd.uchicago.edu.