Abstract
Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD) are caused by mutations of the MAPK pathway, most often BRAFV600E, in myeloid dendritic cells that lead to some overlapping and other unique presentations of the two diseases. LCH occurs in both children and adults, but ECD is primarily found in the latter. The challenges in diagnosing these conditions relates to the rarity of the conditions and that they mimic diseases that are more widely understood, such as certain rashes; bone, lung, and renal diseases; and other malignancies. The histopathology of LCH is definitive, but not so for ECD. Treatment with BRAF and MEK inhibitors has become one of the important advances in the care of these patients.
Learning Objectives
Have a better understanding of the pathophysiology of Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD)
Be aware of new therapies for LCH and ECD
LCH case report
An ear, nose, and throat surgeon has referred a 37-year-old woman with a recent history of chronic otitis externa and a mass in her right external auditory canal. The mass was biopsied and was found to be Langerhans cell histiocytosis (LCH). She also has a history of chronic ulcerative lesions of the vulva. The BRAFV600E mutation was found in the tumor tissue (Table 1).
What is important in her past history? What evaluations should be done? What are the treatment options?
This patient represents the chronic course of LCH for many patients, although only 10% of my adult patients with LCH had the disease in childhood. She also is a good example of the many sites involved with LCH and the need to ask specific questions in history taking (Table 2).
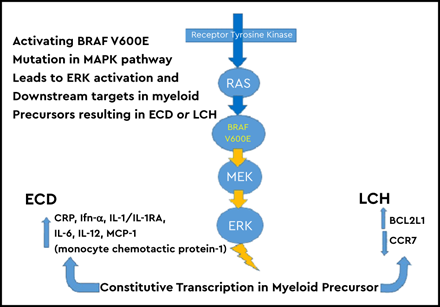
What is the pathophysiology of LCH? In 2010, two important discoveries overturned decades of mystery about the disease. Although it was known to be a clonal proliferation of histiocytes, the conundrum of “immunologic disease” vs malignancy had persisted. Gene expression array experiments proved that the pathologic histiocytes of LCH were myeloid dendritic cells that originated in the bone marrow, not from the normal cutaneous dendritic cells known as “Langerhans cells.”1 The most highly upregulated genes were those of early myeloid cell development and osteopontin. Osteopontin attracts T cells to a lesion. Also, in 2010, the groundbreaking discovery of the BRAF mutation in >50% of biopsies of patients with LCH was published.2 Although the remaining patients did not have the BRAF mutation, all patients with LCH had elevated phosphorylated extracellular signal-regulated kinase (phospho-ERK), meaning that there was some abnormality of the MAPK or associated pathways in every patient with LCH. Subsequent publications showed the BRAF mutation in ≥ 67% of patients with LCH. BRAF-negative patients often have MAP2K mutations, BRAF internal deletions, BRAF rearrangements, and concurrent kinase or other mutations.3-6 Approximately 15% of patients with LCH have no known mutation causing the elevated phospho-ERK. The BRAF mutation proved to be the perfect “bar code” for identifying the LCH cell of origin. Analysis of bone marrow from patients with LCH and in vitro growth of stem cells revealed the BRAF mutation was in CD34+ stem cells, CD11c+ myeloid dendritic cells, CD68+ macrophages, and in rare cases CD3+ T cells. Ultimately, this work led to the concept that LCH is an “inflammatory myeloid neoplasm.” In a mouse model of LCH, upregulation of BCL21, which codes for BCLXL, inhibits apoptosis, so the pathologic CD207+ cells accumulate in the lesion. Activation of the MAPK pathway leads to loss of CCR7 expression, preventing the pathologic cells from migrating out of a lesion.7
Why do patients with LCH have such varied presentations? Why do some with single bone lesions or limited skin lesions resolve spontaneously? Why are some patients cured with a single course of treatment, but others have multiple relapses? The answers to these questions seems to lie in the maturation stage of the myeloid dendritic cell when the BRAF (or other) mutations occurs and whether a patient has the BRAF mutation.
An important clinical division of patients with LCH is whether they have involvement of the liver, spleen, and bone marrow. Patients with those organs involved are classified as “high risk” because in the early Histiocyte Society clinical trials, it was shown that they had the highest risk of a fatal outcome. “Low-risk” patients included those with any other single or multiple non–high-risk organ system affected.8
Bone marrow specimens from high-risk patients had the BRAF mutation in CD34+ stem cells, which could spread to an organ system, but especially the spleen or liver, and remain in the bone marrow.3 It is hypothesized that a BRAF mutation of slightly more mature myeloid dendritic cells would allow circulation to single or multiple organ systems but exclude the high-risk organs. A tissue-resident myeloid dendritic cell with mutated BRAF could account for LCH in a single organ system and limited extent of cutaneous involvement.
If a biopsy of a patient with LCH is BRAFV600E+, what does that tell us about prognosis? A study of 100 patients with LCH found that those with the mutation were more likely to relapse and more likely to have neurodegenerative syndrome if BRAF+ cells were present in the peripheral blood at the time there were no obvious LCH lesions elsewhere.3 There was no clear indication that BRAF mutation in the biopsy differentiated high- and low-risk patients, although circulating BRAF+ cells were almost universally found in the high-risk cases. Subsequently, a larger study reported that a BRAF+ biopsy had a significant association with being a high-risk patient, relapsing, and developing diabetes insipidus and the neurodegenerative syndrome.9 Evaluation of brain specimens from patients with the neurodegenerative syndrome revealed a surprising finding of CD1a+/CD207+/BRAF+ cells in the cerebral blood vessels that migrated into brain tissue and morphed into microglia-like cells staining with CD14+, CD33+, CD163+, P2RY12−/BRAF+.10 Molecular analysis of tissue from the cerebellum and pons, with the most dramatic magnetic resonance imaging (MRI) changes, showed high levels of BRAF+ cells and the osteopontin protein. Brain regions with no MRI abnormalities had essentially no mutated cells or osteopontin.
The LCH case I reviewed highlights many of these comments: multiple organ systems involved and multiple relapses with poor response to vinblastine/prednisone.8 How should she be evaluated now? We perform positron emission tomography (PET) in all patients with new or recurrent LCH to best define sites of disease.11 PET also provides the best indicator of response to therapy. Given the otic involvement, I would perform computed tomography (CT) of the skull to evaluate her mastoids and temporal bones. Patients with diabetes insipidus have a 50% chance of developing infiltrations of the cerebellum, pons, and basal ganglia with LCH cells. About 5% to 10% of these patients will have neurologic deficits, including ataxia, dysmetria, dysarthria, and mental status changes.12 Therefore, MRI of the brain is indicated to define masses of the pituitary and T2-weighted fluid-attenuated inversion recovery imaging abnormalities of the areas I indicated above. Patients with long-standing neurodegenerative symptoms can have abnormalities of the globus pallidus, mesotemporal lobes, and rarely the frontal lobe.13
What are the treatment options for our patient? Vinblastine/prednisone therapy has failed twice. This is not my regimen of choice, because, in a retrospective series at my institution, we found that neuropathy and dislike of steroids were frequent in our adult patients.14 Likewise, the outcomes were poor, with a 30% progression-free survival at 2 years. Other publications have suggested better outcomes, but careful analysis of the data confirms that progression-free survival is not better than 30% to 40%.15 My initial treatment choice for adults with multisystem disease is usually cytarabine 100 mg/m2 for 5 days monthly for 1 year. Our retrospective series revealed 80% progression-free survival at 2 years. Obviously, this should be studied prospectively, but that is unlikely, given the limited patient numbers and other therapy options I discuss further. Cladribine and clofarabine are also used for patients with multisystem LCH.16
This patient’s radiographic examinations revealed no bone lesions and no pituitary or cerebellar abnormalities. She had a soft tissue mass plus skin involvement of the external auditory canal and vulva. I started her on hydroxyurea 500 mg twice daily.17 After 6 months of treatment, all lesions resolved, and we plan to continue the hydroxyurea for another 6 months, then stop. Hydroxyurea alone or with small doses of oral methotrexate has been a successful treatment of the majority of patients with cutaneous LCH I see. Alternatives include oral methotrexate with or without 6-mercaptopurine, thalidomide, or lenolidomide.18,19 Cytarabine was not chosen for this patient, because she had only cutaneous lesions with no bone involvement or evidence of pituitary mass. If I had been treating the patient when she was 23 years old, when she developed diabetes insipidus, then I would have treated her with cytarabine. She did not have circulating BRAFV600E+ cells in her blood, so dabrafenib would not have been chosen. Even if she had had these mutated cells, I would have used hydroxyurea first for economic reasons and because of the fact that the inhibitors never “kill the clone.”
BRAF/MEK inhibitor treatments have been used with increasing frequency in patients with relapsed disease and as initial therapy for those with the neurodegenerative syndrome.20,21 A majority of patients will experience relapse when taken off treatment.
Erdheim-Chester disease
Case report
A nephrologist calls you about a 55-year-old man in renal failure who has some strange tumor around his kidneys and proximal ureters. The biopsy shows lipid-laden macrophages that do not have the atypia of lymphoma cells and stain with anti-CD68 but not CD1a antibodies. The referring physician is mystified because his pathologist said the biopsy was consistent with xanthogranuloma or Erdheim-Chester disease (ECD).22
When you first visit with the patient, you are struck by the exuberant xanthelasmas under his eyes and the history of increasing pain in his distal femurs and proximal tibias for years. In the past year, he has developed chronic headaches and cognitive impairment. You order PET/CT and find the following:
Marked uptake in sclerotic lesions of both distal femurs;
Circumferential coating of the aorta; and
Rind around the kidney (“hairy kidney”) and hydronephrosis.
ECD pathophysiology
The biology of ECD and LCH is remarkably similar, and, in fact, sometimes patients develop both diseases. This is because both diseases derive from a myeloid dendritic cell. In the case of ECD, the developmental pathway is more toward the macrophage lineage. Similar to LCH, 80% of patients have mutations in the RAS/RAF/MEK/ERK cellular signaling pathway.6 Less frequent mutations include ALK, NTRK1, KRAS, NRAS, and PIK3C. Patients with ECD with the BRAF mutation are more likely to have right atrial pseudotumors, cardiac and aortic infiltrations, and pericardial and central nervous system (CNS) involvement.24-26 The proliferation rate as judged by Ki67 staining is low. It is not surprising that a subset of patients have both LCH and ECD, with ECD following or coincident with the LCH diagnosis, given the common origin of these myeloid dendritic cell disorders.27 Up to 10% of patients with ECD or mixed histiocytosis have myeloid neoplasms, including chronic myeloid leukemia and myeloproliferative and myelodysplastic syndromes.28 There is a robust inflammatory response in ECD leading to elevated C-reactive protein in 80% and elevated interferon-γ, interleukin-1 (IL-1)/IL1-RA, IL-6, IL-12, monocyte chemoattractant protein-1, and chemokine ligand 18. The latter has been associated with an exuberant fibroblastic response.29,30
Our patient manifested several of the classic criteria of ECD. Bone involvement occurs in 80% to 95% of patients but is symptomatic in slightly more than one-third. It can be detected by plain radiography, CT, MRI bone scan, or PET scan, usually in the distal femurs and proximal tibias.31 Cardiovascular findings include the “coated aorta” in 40%, which is asymptomatic and not associated with dilatation, dissection, or aneurysm.24 Less than 25% have coronary artery infiltration leading to stenosis and myocardial infarction. Pericardial infiltrates may cause effusions, tamponade, and death. Right atrial pseudotumors are found in slightly over one-third of patients by MRI. Nearly 50% have decreased right ventricular/atrioventricular anatomy, and >60% have decreased right atrial closure.
Pulmonary ECD results in an interstitial lung pattern, interlobular septal thickening, and rarely nodules and ground-glass opacities in one-third to one-half of patients.32 Few patients have pulmonary symptoms.
Hydronephrosis from sheathing of the proximal ureter by the tissue that surrounds the kidney (hairy kidney sign) and retroperitoneal fibrosis are frequent findings.33 Like LCH, ECD can cause diabetes insipidus in nearly one-fourth of patients and frequent anterior pituitary deficiencies of which growth hormone deficiency and hyperprolactinemia or low follicle-stimulating hormone and luteinizing hormone levels are found.34 Unlike patients with LCH, males with ECD may have infiltration of the adrenal glands and testicles.35
CNS damage from ECD occurs in over one-third of patients, frequently in conjunction with xanthelasma and diabetes insipidus. MRI findings are reminiscent of LCH with tumor masses and neurodegenerative findings.36
Pegylated α-interferon has been considered the primary therapy for patients with ECD who have bone, skin, renal, and/or sheathing of the aorta.37 Alternatives include anakinra,38 infliximab,39 and sirolimus + steroids.40
The outcomes for patients with ECD have dramatically improved since the discovery of mutations in the MAP2K pathway and use of specific drugs inhibiting the effects of these specific mutations.41 Some experts recommend reserving the inhibitor therapies for patients with cardiac and CNS involvement and using pegylated interferon as the first intervention.37 Given that nearly 90% of patients with ECD responded to MEK inhibition by cobimetinib, it is likely that more patients will be treated with this class of drugs.21 Principal side effects include cutaneous and cardiac rhythm abnormalities. The majority of patients experience relapse when taken off any inhibitor treatment, so long-term treatment is required.38 We have now reached a consequential level of understanding of both LCH and ECD. It is fascinating that the biology and therapy of two diseases that formerly seemed so distinct are now woven together. It is critical that a biopsy be promptly performed to distinguish LCH and ECD from lymphomas; Rosai-Dorfman disease; and other, more common conditions.
Conflict-of-interest disclosure
K.M. is on the medical advisory board of Sobi.
Correspondence
Kenneth McClain, Baylor College of Medicine, 6701 Fannin St, Suite 1510, Houston, TX 77030; e-mail: klmcclai@txch.org.