Abstract
MRD technologies increase our ability to measure response in acute myeloid leukemia (AML) beyond the limitations of morphology. When applied in clinical trials, molecular and immunophenotypic MRD assays have improved prognostic precision, providing a strong rationale for their use to guide treatment, as well as to measure its effectiveness. Initiatives such as those from the European Leukemia Network now provide a collaborative knowledge-based framework for selection and implementation of MRD assays most appropriate for defined genetic subgroups. For patients with mutated-NPM1 AML, quantitative polymerase chain reaction (qPCR) monitoring of mutated-NPM1 transcripts postinduction and sequentially after treatment has emerged as a highly sensitive and specific tool to predict relapse and potential benefit from allogeneic transplant. Flow cytometric MRD after induction is prognostic across genetic risk groups and can identify those patients in the wild-type NPM1 intermediate AML subgroup with a very high risk for relapse. In parallel with these data, advances in genetic profiling have extended understanding of the etiology and the complex dynamic clonal nature of AML, as well as created the opportunity for MRD monitoring using next-generation sequencing (NGS). NGS AML MRD detection can stratify outcomes and has potential utility in the peri-allogeneic transplant setting. However, there remain challenges inherent in the NGS approach of multiplex quantification of mutations to track AML MRD. Although further development of this methodology, together with orthogonal testing, will clarify its relevance for routine clinical use, particularly for patients lacking a qPCR genetic target, established validated MRD assays can already provide information to direct clinical practice.
Learning Objectives
Update the overview of available AML MRD assays, including newer technologies
Understand current evidence and recommendations for the use of AML MRD assays in clinical practice
Understand key technical aspects for selection, standardization, and interpretation of different AML MRD assays
Case presentation
A 62-year-old man with a normal white blood cell count was diagnosed with cytogenetically normal acute myeloid leukemia (AML), and subsequent molecular studies for European Leukemia Network (ELN) 2017 good and adverse risk mutations, including mutated NPM1, were negative. He was fit enough to be treated with 2 courses of induction chemotherapy (standard UK National Cancer Research Institute [NCRI] AML treatment). Measurable residual disease (MRD) response was assessed by flow cytometry. He achieved a morphological complete remission (CR) but was MRD positive by flow cytometry (at 0.12%) after the second cycle. Because of deterioration in performance status, his comorbidities, and preference, further treatment options did not include transplant. He was known to have IDH1 R132 and DNMT3A mutations and was considered for novel regimens or azacitidine. Should he be monitored if he has further treatment and how?
Introduction
AML is a disease that consists of multiple genotypes with interleukemic, as well as intraleukemic, heterogeneity that is influenced by treatment. Therefore, it is unsurprising that a range of MRD biomarkers and assay platforms have been generated and are relevant to enable personalized AML MRD monitoring.1 However, this contributes to the challenge that MRD testing in 2019 continues to present (ie, deciphering how, when, and even whether it should apply to individual patients). From a survey in 2016-2017, 69% of U.S. leukemia physicians reported routine use of AML MRD, most commonly flow cytometry, followed by polymerase chain reaction (PCR) for mutated NPM1, but only 21% had implemented serial PCR for longer-term monitoring.2 For those not incorporating MRD testing into patient management, cited reasons were lack of resources and uncertainty regarding the use of the results. Of note was the extent to which MRD-directed decision-making varied in hypothetical clinical scenarios. When asked about MRD test positivity postinduction in a patient being considered for transplant, responses were divided equally among recommending against a transplant, additional chemotherapy, and changing the conditioning regimen. This variability reflects the paucity of high-quality evidence currently available to inform such AML MRD–based decisions. It also highlights the need for a more nuanced approach to MRD test interpretation, taking into account that the available evidence from MRD testing differs between AML subtypes. For example, in younger patients with mutated-NPM1 AML, postinduction MRD in the blood by quantitative polymerase chain reaction (qPCR) predicts outcome independently of other mutations, including FLT3,3 and benefit from transplant (hazard ratio = 0.25 for overall survival).4 However, in wild-type NPM1 intermediate-risk AML (as for the patient in the above clinical case) or in older patients, although MRD positivity is also prognostic of poor outcomes,5-7 the effect of additional intensified chemotherapy or transplant requires further evaluation.
It is perhaps easy to forget, with the natural enthusiasm for the prospects of novel MRD technologies, that the well-tested MRD platforms (ie, qPCR and flow cytometry) can provide sophisticated information for MRD levels in most patients, identifying those most likely to relapse with current treatment schedules across all AML risk groups. Clinical trials have assimilated these methods in real time, testing the effect of intensification, early intervention, and novel approaches to improve prognosis or evaluating MRD as an early surrogate for therapeutic efficacy. Outcome data from these trials, together with amalgamated experience in routine clinical practice, stepwise informs how to deal with the results of MRD tests to best help AML patients. In parallel, technical evolution and insights from leukemia biology, together with collaborative efforts for standardization, continuously progress MRD detection. This review focuses on the more recent information from MRD testing, including assay limitations and prospects, that, together with consensus recommendations, can guide current implementation.
Recent guidance from ELN and the U.S. Food and Drug Administration
Since 2018, guidance for the expanding application of MRD in AML has been proposed by 2 organizations. The ELN, through international collaboration, published a consensus document from expert AML MRD laboratories with recommendations for flow cytometric and molecular MRD assays in clinical practice.7 This addressed some of the key factors for MRD measurement, including sampling, recommended approaches, time points, and thresholds of positivity. Perhaps as importantly, the document highlighted areas requiring further work programs to harmonize and progress assays for which efforts by participating laboratories are ongoing. The U.S. Food and Drug Administration (FDA), after a series of workshops over several years, has invited comments on its draft guidance for the use of MRD as a biomarker in regulatory submissions for hematological cancers.8 In it, the FDA lists criteria for MRD assays (eg, that reporting MRD-negative results requires information on detection limits and, perhaps more debatable for certain assays, sensitivity should be ≥1 log below the cutoff for MRD positivity). Referring to AML, the guidance states that each selected MRD marker(s) should reflect the leukemia and not underlying clonal hematopoiesis (false positives). Additionally, data should be provided for the false-negative rate that might result from relapse from a marker-negative clone. The FDA guidance states that MRD could be used to stratify or enrich trial populations, a strategy applied for the RELAZA2 trial9 ; however, when MRD is a trial end point, any patient with missed MRD samples should be categorized as unresponsive in the analysis.
MRD assay considerations
As highlighted by the ELN and FDA articles, there are several important factors to consider when selecting the most appropriate of the current MRD assays and then interpreting results to guide clinical decisions for individual patients. These are the “S” factors:10 specificity, sensitivity of the MRD marker(s), and its stability during AML progression. Further considerations are the context of AML subtype and, if known, associated relapse kinetics, sample type, stage taken, and extent of assay standardization. Another “S,” price ($), is a practical inclusion on the list. Finally, the evidence for correlation with outcome in clinical studies is critical. Preferably, this clinical validation would include MRD testing in real time, as in clinical practice, and would be reproducible by other centers.
Specificity: true vs false MRD positives
Perfect specificity for an assay has been defined as the ability of a method to assess unequivocally the analyte in the presence of other components that are expected to be present. For AML MRD, the analyte is a biomarker of acute leukemia; however, whether the presence of this analyte results in relapse will depend on the test time point, subsequent interventions, and the competing risk of death from other causes. The “other components” consist of any cells/genetic material that are not acute leukemia, as well as artifactual assay background (such as nonspecific antibody binding and autofluorescence in flow cytometry or nonspecific primer binding in PCR). For example, MRD assays by flow cytometry and WT1 RNA overexpression by qPCR (both applicable in the majority of AML patients) have intermediate specificity,, primarily as the result of analyte-type signals (aberrant immunophenotypes or WT1 transcription activity) from normal cells, especially in regenerating marrows.
Acute promyelocytic leukemia (APL) is the paradigm for MRD monitoring in AML. The APL genetic driver from PML-RARA fusion (>95% of APLs) also provides the molecular MRD target of PML-RARA transcripts11 ; this is highly specific, because only treatment-resistant APL cells, including those that may, in time, be relapse initiating, will have this MRD marker. Similarly, other AML subtypes, including core-binding factor (CBF) (RUNX1-RUNX1T1, CBFB-MYH11 fusions) and mutated-NPM1 AMLs, have main genetic drivers that generate specific molecular MRD targets. These analytes are typically representative of the residual AML independently of coexisting mutations but, additionally, are stable markers during AML progression and, thus, strong predictors of relapse by longer-term qPCR MRD monitoring.3,7,12,13 However, even for these, genetic evolution is a consideration for interpretation.14 Mutated-NPM1 AMLs may infrequently relapse as wild-type NPM1, albeit usually later (median of 43 months in a recent study), with associated coexisting and persisting clonal hematopoietic mutations, including DNMT3A.15
It is now apparent that clonal hematopoiesis of indeterminate potential (CHIP)-associated mutations, including the DNMT3A point mutation in the above clinical case, although frequent and stable in AML, can persist posttreatment at high levels, despite longer-term disease-free survival.16-19 Therefore, these are insufficiently specific MRD markers for AML relapse, substantiating the FDA statement that a marker selected to assess MRD should “not reflect underlying clonal hematopoiesis.”8 However, this does not preclude the future utility of MRD assays tracking certain of these mutations, as has already been tested for IDH mutations,20,21 particularly for the efficacy evaluation of appropriate targeted therapies, such as IDH1/2 inhibitors,22,23 a treatment option in our clinical case. Moreover, clonal hematopoietic mutations that cooperate in the progression to acute leukemia could conceivably be monitored in the future as specific biomarkers for preleukemic activity of novel agents. Of particular interest in this regard are CHIP mutations in DNA damage-response genes (eg, TP53 and PPM1D) that are enriched after cytotoxic therapy and associated with an increased risk for developing leukemia.24 Allelic burdens of DNMT3A, TET2, and ASXL1 mutations, including as circulating tumor DNA, may also have potential utility postallogeneic stem cell transplantation to track ablation of patient clonal hematopoiesis.25,26
Sensitivity: true vs false MRD negatives
With the prerequisite of an adequate representative sample, sensitivity for MRD assay targets ranges from 10−2 (current next-generation sequencing [NGS] mutation profiling) to 10−5 to 10−6. The latter is achieved by the established qPCR assays for which the target has high transcript expression (such as NPM1 exon 12 insertion mutations). Current MRD assays cannot test for AML eradication, but reduction of MRD target below the lower limit of detection/quantification at treatment time points (complete remission without minimal residual disease, CRMRD− by ELN criteria27 ) indicates AML clearance of up to 4 logs greater depth than morphology. Not surprisingly, this significantly improves prognostic discrimination in patient cohorts for survival, as well as relapse. Younger adults in CR or CR with incomplete blood count recovery (CRi) after their first course of induction in the NCRI AML17 trial had a 5-year survival of 52%; if also categorized as CRMRD− by flow cytometry (sensitivity of 10−4), survival increased to 63% overall and to 70% when excluding poor-risk patients.5 Observed clinical false negatives (by relapse frequency) from single MRD assessment time points are 20% to 30% in good- or intermediate-risk patients in multiple MRD studies, even for the most sensitive MRD assays.3,28 Monitoring at several time points, such as after each chemotherapy cycle and when applicable sequentially from end of treatment (such as in the Figure 1 schema), captures more information and, consequently, reduces false negatives.5,7,12,13
![(A) Suggested management algorithm for patients with AML with a molecular MRD target. [1] ELN favorable risk patients with <4-log reduction in NPM1-mutant transcripts after first induction are shown to benefit from a CR1 allograft,4 and any positivity in the peripheral blood (PB) after second induction is associated with a very high risk for relapse.3 [2] MRD positivity > 200 copies per 105ABL (ie, molecular persistence) and serially increasing transcript levels after treatment (ie, molecular progression) reliably predict relapse.7 [3] At the end of treatment, patients with CBF AML with high or serially increasing transcript levels are destined to relapse (relevant thresholds are >500 copies [per 105ABL] of RUNX1/RUNX1T1 in the BM or >100 copies in the PB, and > 50 copies of CBFB/MYH11 in the bone marrow [BM] or >10 copies in the PB).7 Salvage according to (C) should be considered for these patients, although there is no evidence that this improves outcome. Conversely, patients with low copy numbers below these thresholds can be safely monitored according to (B). [4] Although CBF patients with an early unfavorable MRD response have a higher risk for relapse, there is insufficient evidence to warrant treatment change7; however, this may prompt initiation of an early donor search. Salvage may be considered in cases with extremely poor early response or if there is an increase while on treatment (ie, molecular progression). [5] Although there is no evidence that standard-risk patients who remain MRD positive benefit from transplant, this is a reasonable approach and is adopted in the current NCRI AML19 and MyeChild01 protocols. (B) Suggested algorithm for sequential monitoring after treatment. Patients with conversion to MRD positivity, confirmed on a second sample with >1-log rise, should be diagnosed with molecular relapse and treated as shown.7 (C) Possible peri-transplant management strategy. [1] Patients with an NPM1 mutation without FLT3 ITD who have transcript levels below 1000 copies in the BM or 200 copies in the PB have a very good outcome after allograft, it is uncertain whether these patients benefit from salvage chemotherapy.89 [2] Patients with high levels of MRD after salvage, without an adequate response to donor lymphocyte infusion (DLI), as well as those to whom these standard therapies cannot be given should be considered for investigational approaches. Figure by Richard Dillon, NCRI Group.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2019/1/10.1182_hematology.2019000060/5/m_hem2019000060cf1.png?Expires=1769904149&Signature=NyLzXA4re06FDTkt7boetEXWaADnH3f2kuIn37cMR1Q98JnUBVa0BdFhp17DTNSsM~dDvxzmq46hxsxfDT3l3d5U4kMBqPVV0iQeAZtR-H7pR6rNTQUd2Ze8xR39Z2SqH4mcvXX~5E4mUWHwNDOz~xXgiqI0kKryXMUYejIR6ZgJW8aj4BGC0F4Q~89VjIgS8oyjuVP2ttttglpVMjCEUyU4npDFp7umGcFL1siBIN-O1BOh7RMXl29bEWz~B6Ns2XVXbI0F2X~VPWCL-InTfWRPTE0B-yCCu4HV5e-kRZbiTZ~qw3K8Z3WHEZYDXYvdE~q8-yYfKoksTdOFARuBZw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Suggested management algorithm for patients with AML with a molecular MRD target. [1] ELN favorable risk patients with <4-log reduction in NPM1-mutant transcripts after first induction are shown to benefit from a CR1 allograft,4 and any positivity in the peripheral blood (PB) after second induction is associated with a very high risk for relapse.3 [2] MRD positivity > 200 copies per 105ABL (ie, molecular persistence) and serially increasing transcript levels after treatment (ie, molecular progression) reliably predict relapse.7 [3] At the end of treatment, patients with CBF AML with high or serially increasing transcript levels are destined to relapse (relevant thresholds are >500 copies [per 105ABL] of RUNX1/RUNX1T1 in the BM or >100 copies in the PB, and > 50 copies of CBFB/MYH11 in the bone marrow [BM] or >10 copies in the PB).7 Salvage according to (C) should be considered for these patients, although there is no evidence that this improves outcome. Conversely, patients with low copy numbers below these thresholds can be safely monitored according to (B). [4] Although CBF patients with an early unfavorable MRD response have a higher risk for relapse, there is insufficient evidence to warrant treatment change7 ; however, this may prompt initiation of an early donor search. Salvage may be considered in cases with extremely poor early response or if there is an increase while on treatment (ie, molecular progression). [5] Although there is no evidence that standard-risk patients who remain MRD positive benefit from transplant, this is a reasonable approach and is adopted in the current NCRI AML19 and MyeChild01 protocols. (B) Suggested algorithm for sequential monitoring after treatment. Patients with conversion to MRD positivity, confirmed on a second sample with >1-log rise, should be diagnosed with molecular relapse and treated as shown.7 (C) Possible peri-transplant management strategy. [1] Patients with an NPM1 mutation without FLT3 ITD who have transcript levels below 1000 copies in the BM or 200 copies in the PB have a very good outcome after allograft, it is uncertain whether these patients benefit from salvage chemotherapy.89 [2] Patients with high levels of MRD after salvage, without an adequate response to donor lymphocyte infusion (DLI), as well as those to whom these standard therapies cannot be given should be considered for investigational approaches. Figure by Richard Dillon, NCRI Group.
(A) Suggested management algorithm for patients with AML with a molecular MRD target. [1] ELN favorable risk patients with <4-log reduction in NPM1-mutant transcripts after first induction are shown to benefit from a CR1 allograft,4 and any positivity in the peripheral blood (PB) after second induction is associated with a very high risk for relapse.3 [2] MRD positivity > 200 copies per 105ABL (ie, molecular persistence) and serially increasing transcript levels after treatment (ie, molecular progression) reliably predict relapse.7 [3] At the end of treatment, patients with CBF AML with high or serially increasing transcript levels are destined to relapse (relevant thresholds are >500 copies [per 105ABL] of RUNX1/RUNX1T1 in the BM or >100 copies in the PB, and > 50 copies of CBFB/MYH11 in the bone marrow [BM] or >10 copies in the PB).7 Salvage according to (C) should be considered for these patients, although there is no evidence that this improves outcome. Conversely, patients with low copy numbers below these thresholds can be safely monitored according to (B). [4] Although CBF patients with an early unfavorable MRD response have a higher risk for relapse, there is insufficient evidence to warrant treatment change7 ; however, this may prompt initiation of an early donor search. Salvage may be considered in cases with extremely poor early response or if there is an increase while on treatment (ie, molecular progression). [5] Although there is no evidence that standard-risk patients who remain MRD positive benefit from transplant, this is a reasonable approach and is adopted in the current NCRI AML19 and MyeChild01 protocols. (B) Suggested algorithm for sequential monitoring after treatment. Patients with conversion to MRD positivity, confirmed on a second sample with >1-log rise, should be diagnosed with molecular relapse and treated as shown.7 (C) Possible peri-transplant management strategy. [1] Patients with an NPM1 mutation without FLT3 ITD who have transcript levels below 1000 copies in the BM or 200 copies in the PB have a very good outcome after allograft, it is uncertain whether these patients benefit from salvage chemotherapy.89 [2] Patients with high levels of MRD after salvage, without an adequate response to donor lymphocyte infusion (DLI), as well as those to whom these standard therapies cannot be given should be considered for investigational approaches. Figure by Richard Dillon, NCRI Group.
Sample considerations for MRD interpretation
It is a sine qua non that a good quality bone marrow (BM) is most likely to be representative of residual AML for the majority of patients without extramedullary disease, hence, the present recommendation of a first-pull BM for almost all AML MRD assays to reduce false negatives from suboptimal sensitivity. Leukocyte numbers, cell viability (affected by transit time), hemodilution, and hypoplasia all contribute to limiting the sensitivity/lower level of quantification for any BM sample, independently of the theoretical assay sensitivity. Flow cytometric assays can assess and should incorporate information on all of these factors in the MRD report. MRD negativity from an antibody combination testing 250 000 leukocytes will only reach a detection limit of 0.01% (with a 20% coefficient of variation [CV]). Molecular assays include housekeeping gene (ABL) copies as control for nucleated cell numbers, but current assays cannot differentiate hemodilution and cell type unless performed on presorted cells. Single-cell assays in the future may be able to combine phenotype, as well as RNA expression and mutation profile,29 but these currently are very expensive and can only evaluate small cell numbers.
Is blood informative for AML MRD?
Despite blood (peripheral blood [PB]) providing lower sensitivity than BM (1 log less for mutated NPM13 ), measuring mutated-NPM1 transcripts in PB postinduction is highly prognostic for the mutated-NPM1 subgroup.3,4 Interestingly the “false-negative” relapse risk is not increased for MRD negativity in blood vs BM.4 This implies that, in the ∼25% of mutated-NPM1 patients with only BM positivity postinduction, the MRD is at a level concomitant with clearance by consolidation or is from nonleukemic-initiating more mature BM cells. However, BM is recommended for maximal sensitivity during later sequential monitoring, because BM positivity by qPCR targets usually precedes that of blood, providing an increased time window for any interventions.7
Reduced sensitivity may be compensated for by a differential increase in specificity when testing blood for flow cytometric MRD30 (because of fewer normal progenitors/precursors and, therefore, less “noise”), as observed when measuring WT1 levels.31 Relapse-free survival32 and overall survival30,33 appear to be significantly better for patients with MRD-negative blood samples postinduction and postconsolidation in heterogeneous smaller cohorts. These encouraging results merit further evaluation in older and intermediate/poor-risk AML patients, most of whom lack sensitive qPCR molecular markers. There is also preliminary evidence that PB clonal profiles may be representative of BM during treatment,34 as well as at diagnosis, at least for higher frequency mutations. In a small series of decitabine-treated patients, NGS (non–error-corrected) measurements of variant allele frequencies (VAFs) (at >5%) for mutation profiles in PB samples with <60% lymphocytes correlated well with paired BM results.35
How we would monitor: CBF AMLs and mutated-NPM1 AMLs
Patients with CBF or mutated-NPM1 AML, by the nature of their leukemic-specific fusion transcripts (RUNX1-RUNX1T1, CBFB-MYH11) or insertion/deletion mutations (NPM1), can be monitored by qPCR MRD assays that combine high sensitivity with specificity and are extensively validated. Monitoring schedules (such as in Figure 1) and reporting of results for comparability should be based on ELN guidelines,7 although these are only evidence based for younger patients.36 After treatment completion, qPCR MRD assessments are recommended for ≥2 years at 3-month intervals when the prior result was negative or revealed low copy numbers. Average kinetics from molecular relapse to clinical relapse ranges from >3 to 4 months for CBFB-MYH11 to 2 to 3 months for RUNX1-RUNX1T1 or mutated-NPM1 when FLT3-ITD positive.37 There are no new data to support a survival benefit from preemptive intensification in CBF AMLs, despite the clear association between MRD status (postinduction/consolidation or off-treatment) and clinical progression for younger patients.7,38 Published studies indicate that allogeneic transplantation can be avoided in FLT3-ITD–positive mutated-NPM1 younger adults when these patients remain in complete molecular first remission following standard induction,7 but it is uncertain whether concomitant treatment by Flt3 inhibitors alters relapse kinetics and, therefore, optimal off-treatment sampling intervals.
qPCR assays have been developed for other fusion transcripts, such as from rearrangements involving KMT2A (MLL), NUP98, and NUP214. Although these can track response (Figure 1), there are insufficient data to provide guidance for their use; hence, it is recommended to also assess response by flow cytometric MRD postinduction.7
What about patients with FLT3 internal tandem duplications?
Given the frequent prevalence in AML patients, the high probability and rapid kinetics of relapse, and the continued development of effective targeted therapy, there is great interest in monitoring of AML clones containing FLT3 internal tandem duplications (FLT3-ITDs). FLT3-ITD mutations represent late events in leukemic development and, hence, are not always detectable at relapse, particularly after targeted therapy; they are often “replaced” by another signaling variant (eg, RAS, Kit, or a different FLT3 variant).39,40 Despite the limitation of potential false-negative tests, FLT3-ITD MRD testing has utility, because a positive result in an AML patient otherwise thought to be in remission is highly suggestive of MRD and is associated with a high likelihood of relapse, often with a short lead time.41-43 FLT3-ITD mutations consists of nucleotide sequence inserts of variable length and location between patients, making 1 universal approach to low-level quantitative assessment by conventional PCR and bioinformatic mapping of data from NGS challenging. However, this technical constraint has been mitigated by novel PCR methods44 and sequencing approaches.39,41,43,45 In ∼50% of adult AML cases, FLT3-ITD mutations will co-occur with a more stable AML MRD marker, such as mutated NPM1 or t15:1746 ; therefore, it is recommended to also track such markers,7 particularly for patients receiving FLT3-ITD–directed therapy. For those without a mutation other than FLT3-ITD to track, the expression of WT1 is known to be elevated in these patients,31,47 although the use of this AML MRD target for routine testing remains controversial. Flow cytometric MRD detection is recommended for response evaluation and monitoring for those patients with AML that cannot be tracked by a validated qPCR MRD assay.
Newer molecular technologies in AML MRD detection
The past decade of focus on cancer genomics has elucidated the genetic basis of AML and provided a wide range of molecular targets suitable for new drug development, as well as potentially for leukemic disease burden tracking. Although such approaches are highly publishable, several limitations prevent direct translation to the clinic at present. Unlike conventional quantitative PCR (Figure 2A), digital PCR, developed in the 1990s, allows absolute quantification of abnormal DNA sequences by partitioning each template molecule for the PCR reaction into an individual compartment (Figure 2B). Advantages of this technology include high sensitivity as the result of a low background error rate, highly accurate quantification due to elimination of template competition, limited bioinformatic requirements, and a rapid “sample-to-result” turnaround time.28 Disadvantages include the need to develop and validate assays for each individual target sequence, limited multiplexing capacity, and the inability to perform discovery on serial samples (eg, to detect selection of an independent TP53-mutated clone during therapy), making it unsuitable to detect relapses associated with clonal evolution. This technology is likely to be most useful for orthogonal validation of other technology and for tracking of common highly conserved “hotspot” variants, such as those seen in NPM1 and IDH1/IDH2 genes.20,48
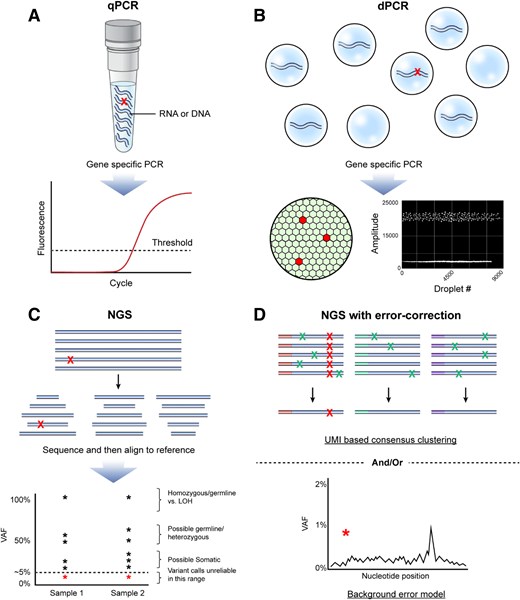
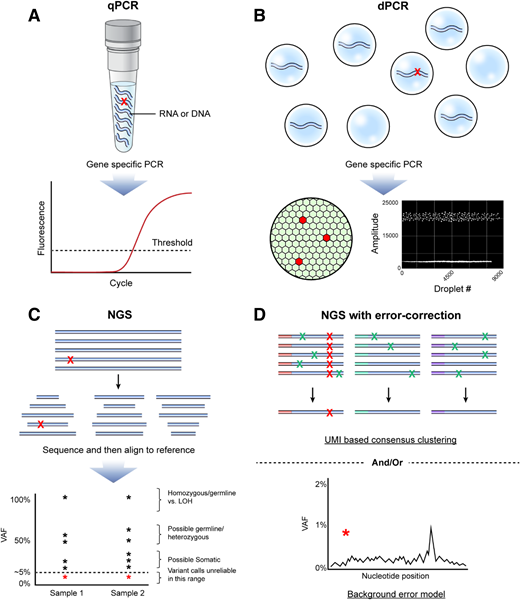
(A) qPCR is a common method for quantification of nucleic acid with real-time monitoring of the amplification of target of interest (eg, variant sequence shown with red X). Advantages include ubiquitous presence in most clinical laboratories, fast turnaround time, high sample throughput, and broad dynamic range. Disadvantages include limited number of suitable targets/assays available, relative lack of multiplexing ability, need to validate each target/assay individually, potential for false-negative results because of sample impurity, and limited ability to accurately discriminate between very low levels of target as seen in MRD. (B) Rather than performing the PCR reaction in “bulk,” digital PCR partitions the template of interest into individual compartments (top), improving the performance compared with qPCR because of the lower background error rate (lower right), elimination of template competition, and digital result output, allowing absolute quantification (lower left). Lack of deep multiplexing ability and the need to validate each target/assay individually remain limitations. (C) NGS has revolutionized the initial clinical diagnostic evaluation of AML by allowing for simultaneous evaluation of multiple target regions typically selected from those known to be often mutated in AML. NGS is useful for discovery of mutations present in the range from 5% to 100% of a sample (VAF). However, not all variants detected will be pathogenic somatic mutations, and care should be taken to consider the possibility of identification of homozygous or heterozygous germline variants, as well as loss-of-heterozygosity (LOH) events. Variant discovery below a VAF of 5% using panels designed for profiling variants at diagnosis is challenging because of the lack of sensitivity and high false-positive rates. Red asterisks represent low-level variant calls that should be regarded with particular caution as within the range of background error for conventional NGS. (D) NGS for AML MRD performed in recent high-quality research studies has typically included error correction (upper), by incorporation of UMIs, followed by consensus determination of true (red X) variants vs false positives introduced by the technique (green X) and/or bioinformatic approaches to model background error rates at each nucleotide position in those not having a variant and determine the probability that the observed variant is a true positive (red asterisk) (lower). Figure by Erina He, National Institutes of Health Medical Arts.
(A) qPCR is a common method for quantification of nucleic acid with real-time monitoring of the amplification of target of interest (eg, variant sequence shown with red X). Advantages include ubiquitous presence in most clinical laboratories, fast turnaround time, high sample throughput, and broad dynamic range. Disadvantages include limited number of suitable targets/assays available, relative lack of multiplexing ability, need to validate each target/assay individually, potential for false-negative results because of sample impurity, and limited ability to accurately discriminate between very low levels of target as seen in MRD. (B) Rather than performing the PCR reaction in “bulk,” digital PCR partitions the template of interest into individual compartments (top), improving the performance compared with qPCR because of the lower background error rate (lower right), elimination of template competition, and digital result output, allowing absolute quantification (lower left). Lack of deep multiplexing ability and the need to validate each target/assay individually remain limitations. (C) NGS has revolutionized the initial clinical diagnostic evaluation of AML by allowing for simultaneous evaluation of multiple target regions typically selected from those known to be often mutated in AML. NGS is useful for discovery of mutations present in the range from 5% to 100% of a sample (VAF). However, not all variants detected will be pathogenic somatic mutations, and care should be taken to consider the possibility of identification of homozygous or heterozygous germline variants, as well as loss-of-heterozygosity (LOH) events. Variant discovery below a VAF of 5% using panels designed for profiling variants at diagnosis is challenging because of the lack of sensitivity and high false-positive rates. Red asterisks represent low-level variant calls that should be regarded with particular caution as within the range of background error for conventional NGS. (D) NGS for AML MRD performed in recent high-quality research studies has typically included error correction (upper), by incorporation of UMIs, followed by consensus determination of true (red X) variants vs false positives introduced by the technique (green X) and/or bioinformatic approaches to model background error rates at each nucleotide position in those not having a variant and determine the probability that the observed variant is a true positive (red asterisk) (lower). Figure by Erina He, National Institutes of Health Medical Arts.
DNA sequencing, typically consisting of “panels” covering the small regions of the genome known to be often somatically mutated in AML patients, has increasingly become part of the standard initial diagnostic workup, given the importance of such mutations in risk stratification and therapy selection27 (Figure 2C). Because detected mutations are thought to originate from the leukemic clone and may persist during remission in patients at an increased risk for relapse,17 it may be tempting to use the same NGS test for AML MRD detection. Unfortunately, as suggested by the absence of recommendations on NGS in the current ELN AML MRD guidelines, most NGS panels currently used at the time of AML diagnosis are not fit for the purpose of MRD detection because of limited sample input, insufficient read-depth, and lack of appropriate error correction (Table 1). NGS “AML panels” at diagnosis are used to determine likely somatic variants typically with a VAF (also known as mutant allele frequency) ≥ 5% (ie, 5 variant reads per every 100 at that position). Accordingly, those tests use genomic DNA inputs as low as 10 ng, which represents <8000 total cells (typical range of such tests, 10-100 ng). Given losses during NGS library preparation, a requirement to detect 3 to 5 unique copies of each variant (each representing a heterozygous mutation from a unique cell) to call MRD, and Poisson distribution sampling considerations at the limit of detection (such that the FDA guidance recommended the MRD threshold be 10-fold higher than the theoretical limit of detection), it is understandable that most high-quality AML MRD NGS research studies have used DNA inputs in the 200- to 500-ng range (ie, ≥1 mL of marrow aspirate or blood). Similarly, most AML MRD approaches using NGS, in recognition of the massive number of false-positive results, particularly for single nucleotide variants when considering potential variants < 2% VAF, have used some form of error correction in the form of laboratory techniques (eg, use of unique molecular identifiers [UMIs]), followed by consensus clustering and/or bioinformatic techniques, including models incorporating nucleotide position-specific background error rates resulting from PCR-based library preparation and sequencing itself17,49 (Figure 2D). The sequencing depth requirement is often substantially higher for AML MRD than the one routinely used for diagnostic testing, because, in addition to the increased sample input, each unique DNA molecule typically will be sequenced 5 or 10 times in UMI-based error-corrected sequencing. Finally, the prognostic significance of AML “MRD,” as detected by NGS, may not be equivalent to that detected by qPCR or flow cytometry; indeed, there is already considerable evidence that detection of some somatic mutations may be more prognostic than others.17,50,51
In contrast, AML MRD targets for qPCR have already been extensively validated and have been approved by the ELN consensus guidelines.7 This recently led to the development of an RNA sequencing assay capable of simultaneously detecting all of these molecular targets in a single standardized NGS workflow while maintaining a limit of detection comparable to qPCR.52 Future NGS AML MRD assays may combine RNA- and DNA-based approaches.
Update for immunophenotypic MRD
The ELN article provides a framework for the harmonization/standardization of flow cytometric MRD detection.7 Consensus guidance in this covers the most reliable of extensively tested markers for tracking leukemic aberrant immunophenotypes, use of 8-color panels (acceptable in clinical laboratories to increase single-cell information), the importance of harmonizing cytometer settings for comparable antibody-stained cell profiles, and some technical recommendations for sample processing. Knowledge of the diagnostic Leukemic Associated ImmunoPhenotypes (LAIP) prevents reporting MRD negativity in the 5% to 10% of marker-negative patients (no identifiable LAIP at diagnosis or at follow-up), whereas a different-from-normal (DfN) analysis can detect phenotypic changes from leukemia evolution.14 Consequently, it is advised to combine information from both diagnostic LAIP and DfN analysis to minimize false negatives. Defining MRD positivity by ≥0.1% of leukocytes (1 log greater than the limit of detection) is proposed, because this threshold is relevant in most studies. However, the proviso for this is that some patients with quantifiable MRD at <0.1% may have residual AML that is prognostic, for example, resulting from MRD underrepresentation by hemodilution, or potentially due to smaller, but more chemo-resistant, amounts after consolidation,53,54 or upfront higher-intensity treatment,55 or in older patients6 . Risk discrimination from low-level MRD may also depend on genetic subgroup; detectable, but <0.1% flow cytometric, MRD after a first induction course was associated with a significantly increased relapse risk in mutated-NPM1 and CBF AMLs but not in wild-type NPM1 intermediate-risk patients.5
Feasibility of harmonization/standardization
Experience from acute lymphoblastic leukemia flow cytometric MRD work has shown that multicenter standardization56 can be achieved with interpretive discordance reduced among experienced laboratories by feedback schemes.57 Recent efforts by some multicenter flow cytometry laboratory networks, such as in Germany and France, are showing that this is also feasible for AML MRD, despite differences in cytometers and so forth. Figure 3 shows an example of an implemented harmonization strategy (Adriana Plesa and Christophe Roumier, on behalf of the Acute Leukemia French Association [ALFA], written communication, 10 May 2019). The ELN is testing a consensus standardized tube as a template to simplify interlaboratory set-up and comparability for multicenter interpretation.58
![(A) Strategy applied for flow cytometric AML MRD multicenter harmonization by the ALFA Intergroup. [1] Rationale of AML MRD flow panel design was based on simplicity, reproducibility, and cost. Tube 1 was a core combination for LAIP detected at diagnosis and/or by different-from-normal analysis, tube 2 was targeted to aberrancies of CD34+CD38− cells (immunophenotypic LSCs), and tube 3 was an optional development tube for monocytic aberrancies. [2] Flow cytometer fluorescent settings were harmonized (“mirrored”) between Canto vs Navios cytometer platforms. Voltages were set to reach target mean fluorescence intensity (MFI) values by acquisition of rainbow calibration beads without compensation for fluorescent channels FL1 to FL8 on the Canto cytometers; these rainbow bead settings were transposed to Navios cytometers by applying MFI target = Canto target/256. Mirrored (superimposable) target peaks for both cytometers are shown for FL1 and FL8 fluorescent channels with an example of resulting comparable antibody profiles between cytometer platforms. (B) [3] Quality controls for reproducibility of staining profiles from harmonized cytometer settings/sample processing between cytometers/laboratories. Examples shown are for CD117 and CD38 expression intensity on CD34+ gated mononuclear cells of tube 1 from 10 shared BM samples stained and then acquired on Canto or Navios flow cytometers. Intensity profiles are similar between cytometer platforms for each sample. [4] External quality assessment for all harmonized steps from preanalytical to final gating analyses by distribution of a normal BM to 22 participating laboratories (cytometer platforms: 12 Cantos, 10 Navios). Example shows that strong reproducibility can be achieved in the detection of rare events (shown for CD34+CD38−) among 22 participating laboratories. Figure by Christophe Roumier and Adriana Plesa, ALFA.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2019/1/10.1182_hematology.2019000060/5/m_hem2019000060cf3.png?Expires=1769904149&Signature=ixd12y82zXI6e-7BGTg2wZE5cnGoIfQjDH7-ekE-9HCBnVH6WEe9bb-qMfQlR2q60cRqVJZxPgk2KFeyQzMDlb4NBotqpd7rxswwm4xWJo4XNrOFT6vm7feLH2dCYmAO5ZrIsNDzqBwT-l9ES7UDsIlcKft8mQne53OgAqiSKtxKI5oPJXvWtkJx3TpdEAgK-~ZVhd0HVbWd1HXPovN2whCzBTtbE6llNsJPSeHggoR7r8W~7kHDTmoxhRjFDfBjXYQ~iS~iR36LazpO-qw3LVNGKQRh8NxWwCRaDSswZLv192sgdVzN1bFtaP~yhOJto2Ym6EnpSrBqRfm7Acm0sQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Strategy applied for flow cytometric AML MRD multicenter harmonization by the ALFA Intergroup. [1] Rationale of AML MRD flow panel design was based on simplicity, reproducibility, and cost. Tube 1 was a core combination for LAIP detected at diagnosis and/or by different-from-normal analysis, tube 2 was targeted to aberrancies of CD34+CD38− cells (immunophenotypic LSCs), and tube 3 was an optional development tube for monocytic aberrancies. [2] Flow cytometer fluorescent settings were harmonized (“mirrored”) between Canto vs Navios cytometer platforms. Voltages were set to reach target mean fluorescence intensity (MFI) values by acquisition of rainbow calibration beads without compensation for fluorescent channels FL1 to FL8 on the Canto cytometers; these rainbow bead settings were transposed to Navios cytometers by applying MFI target = Canto target/256. Mirrored (superimposable) target peaks for both cytometers are shown for FL1 and FL8 fluorescent channels with an example of resulting comparable antibody profiles between cytometer platforms. (B) [3] Quality controls for reproducibility of staining profiles from harmonized cytometer settings/sample processing between cytometers/laboratories. Examples shown are for CD117 and CD38 expression intensity on CD34+ gated mononuclear cells of tube 1 from 10 shared BM samples stained and then acquired on Canto or Navios flow cytometers. Intensity profiles are similar between cytometer platforms for each sample. [4] External quality assessment for all harmonized steps from preanalytical to final gating analyses by distribution of a normal BM to 22 participating laboratories (cytometer platforms: 12 Cantos, 10 Navios). Example shows that strong reproducibility can be achieved in the detection of rare events (shown for CD34+CD38−) among 22 participating laboratories. Figure by Christophe Roumier and Adriana Plesa, ALFA.
(A) Strategy applied for flow cytometric AML MRD multicenter harmonization by the ALFA Intergroup. [1] Rationale of AML MRD flow panel design was based on simplicity, reproducibility, and cost. Tube 1 was a core combination for LAIP detected at diagnosis and/or by different-from-normal analysis, tube 2 was targeted to aberrancies of CD34+CD38− cells (immunophenotypic LSCs), and tube 3 was an optional development tube for monocytic aberrancies. [2] Flow cytometer fluorescent settings were harmonized (“mirrored”) between Canto vs Navios cytometer platforms. Voltages were set to reach target mean fluorescence intensity (MFI) values by acquisition of rainbow calibration beads without compensation for fluorescent channels FL1 to FL8 on the Canto cytometers; these rainbow bead settings were transposed to Navios cytometers by applying MFI target = Canto target/256. Mirrored (superimposable) target peaks for both cytometers are shown for FL1 and FL8 fluorescent channels with an example of resulting comparable antibody profiles between cytometer platforms. (B) [3] Quality controls for reproducibility of staining profiles from harmonized cytometer settings/sample processing between cytometers/laboratories. Examples shown are for CD117 and CD38 expression intensity on CD34+ gated mononuclear cells of tube 1 from 10 shared BM samples stained and then acquired on Canto or Navios flow cytometers. Intensity profiles are similar between cytometer platforms for each sample. [4] External quality assessment for all harmonized steps from preanalytical to final gating analyses by distribution of a normal BM to 22 participating laboratories (cytometer platforms: 12 Cantos, 10 Navios). Example shows that strong reproducibility can be achieved in the detection of rare events (shown for CD34+CD38−) among 22 participating laboratories. Figure by Christophe Roumier and Adriana Plesa, ALFA.
Immunophenotypic leukemic stem cell assays
Weak/negative CD38 expression on CD34+ cells (CD34+CD38−) identifies progenitors that are enriched for functional hematopoietic stem cell activity in normal BM. High frequencies of CD34+CD38− cells in AML at diagnosis (variability shown in Figure 4) have an independent adverse prognostic impact,59-61 consistent with this immunophenotypic subpopulation as a biomarker for leukemic stem cell (LSC)-like chemoresistance in some AMLs. Leukemic CD34+CD38− cells can express aberrant markers62 that provide flow cytometric “LSC” targets with higher assay specificity due to less background from normal counterparts than with bulk progenitors. The approach has been refined and standardized for a “different-from-normal” strategy by construction of a single “LSC” tube that combines multiple CD34+CD38− aberrant “LSC” markers.63 Immunophenotypic quantitation of an immunophenotypic LSC population prior to allogeneic transplant was prognostic for relapse-free survival.64 Moreover, immunophenotypic LSC frequency in CR had significant additive prognostic value to standard MRD by LAIP or mutated-NPM1 qPCR in a large cohort, primarily as a result of increased specificity for very poor outcomes in the ∼10% of patients with positivity for LSC and standard MRD.65 Although increasing assay sensitivity to 1 in a million by testing more cells could reduce the false-negative relapse frequency observed in this study (3-year cumulative incidence of relapse [CIR] of 35% for double LSCneg/MRDneg), weak/negative CD38 may not be an appropriate or stable marker for relapse-initiating LSCs in a subset of patients.66,67
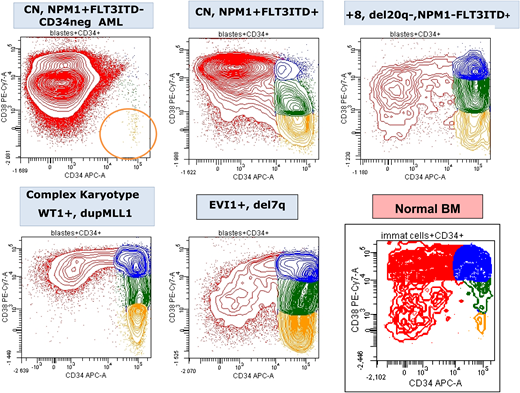
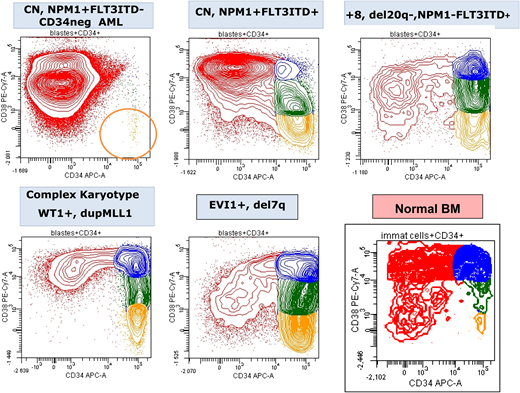
CD34/CD38 expression pattern of blasts from 5 diagnostic AML samples showing the variability in the frequency of the most immature leukemia cells (CD34+CD38−; orange) compared with normal BM. CN, normal cytogenetics. Figure by Adriana Plesa and Christophe Roumier, ALFA.
CD34/CD38 expression pattern of blasts from 5 diagnostic AML samples showing the variability in the frequency of the most immature leukemia cells (CD34+CD38−; orange) compared with normal BM. CN, normal cytogenetics. Figure by Adriana Plesa and Christophe Roumier, ALFA.
Future perspectives in immunophenotypic MRD
Ongoing discovery of leukemic aberrant phenotypes expands and rationalizes the repertoire of informative markers that can inform future routine AML MRD antibody panels,68 as well as immunotherapy targets. For example, IL1RAP has been identified by proteomic66 and single-cell RNA sequencing29 strategies as a discriminatory marker, particularly in FLT3-ITD AML.
High-dimensional immunophenotyping using cytometry by time of flight (CyTOF), when available, allows deeper and/or broader population coverage because of increased parameters (>40) compared with that possible with flow cytometry. When combined with novel data-analysis algorithms,69 CyTOF data can reveal biologically important leukemic and immune cell subpopulations that are missed by traditional approaches. Although CyTOF has proven to be a powerful discovery tool, its slow acquisition rates (500 events per second) preclude applying the technology to clinical laboratory assays that require high cell numbers, including MRD assays. However, because individual samples can be metal barcoded, there may be potential for multiplexing of diagnostic samples, achieving higher throughput and reducing data collection variation. This approach could screen for the most informative markers to guide construction of improved flow cytometric MRD panels or to enable personalized profiling for immunotherapeutic targets. The analysis algorithms developed for high-dimensional immunophenotyping have also been applied to flow cytometric data70 and may, with further development, provide tools for unsupervised or partially unsupervised AML MRD analysis in the future.71
Immunophenotyping vs molecular MRD detection: friend or foe?
Initial evidence points to molecular and flow cytometric MRD providing complementary independent information at the same time points during active treatment, particularly when the molecular assays have lower sensitivity and specificity.17,18,21,72,73 After induction, ∼30% of patients in 2 studies17,18 had discordant results between flow cytometric and standard NGS MRD (excluding the main CHIP mutations), and they had an intermediate risk for relapse (∼50% at 4 years) in the larger cohort.17 To understand how best to improve and deploy these assays, it will be helpful in the research setting to define whether their respective false-positive/negative results cluster in certain AML subtypes and whether there is any overlap. Of particular interest in this regard are AMLs without a sensitive qPCR target, including those with greater clonal complexity, such as the chromatin-spliceosome group.
Flow cytometry vs morphology to assess remission
Flow cytometric assays continue to provide the fastest turnaround time for evaluating blast percentages and MRD so that routine results may be available concurrently with, if not before, morphology reporting. Interobserver variability for morphological BM blast counts has been reported recently as greatest for blast percentages from 2% to 10% (and only moderate agreement for <2% and >10%), whereas reference percentages calculated from digital trephine images correlated well with flow cytometric blast percentages of first-pull BM.74 Evaluation of leukemic aberrant immunophenotypes by MRD antibody panels adds specificity and sensitivity for discriminating leukemic blasts from normal blasts. Adults75,76 and children77,78 who are refractory by morphology, but MRD negative by flow cytometry after first induction, have a good prognosis (60% 3-year survival in 2 adult trial cohorts75 ), equivalent to those in CRMRD-. Also of note is that, in a series of 87 patients morphologically categorized as relapsed, none were MRD negative by flow cytometry.79 Together, these results support refining the present criteria for refractory and relapsed AML to incorporate flow cytometric MRD-negative results when a validated assay is available.
Because MRD positivity in CR and morphologic refractory disease appear equivalent for outcomes preallogeneic transplant53,80 (with the caveat of unavoidable selection bias), does blast enumeration for the criteria of resistant disease and partial remission add prognostic information? In younger adults treated in the NCRI AML17 trial, patients in partial remission (International Working Group criteria) or MRD-positive CR after a first course of induction had an equivalent intermediate prognosis (∼40% 5-year survivals), with the exception of MRD-positive patients with incomplete count recovery, who had a much poorer prognosis (19% 5-year survival). Patients with resistant disease had a similar outcome to the latter group.5 Thus, when flow cytometric MRD is incorporated into response assessment after first induction, the prognostic effect of ≥5% blasts by morphology appears to be restricted to the subgroup with resistant disease.
Treatment changes based on MRD detection
MRD status after induction
In the case of mutated-NPM1 AML,4 and potentially for wild-type NPM1 intermediate-risk AML,5 exploratory analyses support directing allogeneic transplant in first remission only to those patients testing positive for MRD postinduction chemotherapy (by qPCR for mutated-NPM1 or flow cytometry for wild-type NPM1). However, when ELN 2017 adverse-risk patients are included, the benefit from allogeneic transplant as consolidation appears to be at least equivalent between MRD-negative and MRD-positive patients.81,82 This supports allogeneic transplant in first remission as the best approach for adverse-risk patients, even when achieving MRD negativity early in treatment.
The extent to which outcome is altered by intensifying consolidation in those who are MRD positive at the postinduction time point will be further informed by forthcoming data in trials implementing this strategy in younger adults (eg, NCRI AML17/19 and HOVON 132 for ELN intermediate risk) and in older adults (NCRI AML18).
MRD status after consolidation
The CETLAM AML12 and the GIMEMA AML1310 phase 2 trials have investigated adjusting allogeneic transplant allocation by postconsolidation MRD levels in addition to genetic risk. Initial reports from these trials83,84 show the feasibility of real-time MRD treatment stratification and, in the GIMEMA study, disease-free survival was similar between MRD-positive and MRD-negative intermediate genetic risk patients. The low patient numbers in the analyses of the effect of transplant directed by postconsolidation MRD reinforces the difficulty of testing nonbiased transplant interventions.
Monitoring off treatment
A suggested MRD-directed treatment algorithm for AML patients with stable molecular qPCR MRD targets is shown in Figure 1. When patients remain low risk by MRD levels after induction and consolidation, sequential 3-month qPCR MRD monitoring is recommended to allow a greater time window for treatment decisions, because an increase in target transcript levels (as defined by the ELN7 ) precedes clinical relapse.
For AML patients without a qPCR MRD target, the role of off-treatment sequential monitoring requires further evaluation. However, similar to MRD status after induction and consolidation, pretransplant and potentially posttransplant MRD testing can inform the risk of poorer outcomes.
MRD status pretransplant
Although it is clear that persistent MRD positivity in AML patients in morphological remission prior to allogeneic transplant49,53,80,85 is associated with increased relapse and decreased survival after transplant, because of the lack of randomized clinical trials it is unproven whether additional intervention can improve clinical outcomes rather than simply increase treatment toxicity. Results from ultradeep NGS on pretransplant blood of AML patients in the BMT CTN phase 3 randomized trial 0901 were recently presented; in patients with an AML-associated variant detected, increased posttransplant relapse risk and inferior overall survival were noted in those randomized to reduced intensity conditioning compared with those randomized to myeloablative conditioning.86 This study provides the best current evidence that intervention on the MRD state in AML may improve clinical outcomes.
MRD posttransplant
For those patients who develop MRD positivity off treatment, preemptive treatment may be feasible even after allogeneic transplant. In the RELAZA2 phase 2 trial, azacitidine converted 36% of 53 patients back to CRMRD−, with 20% maintaining this status during the 2-year follow-up. Although such nonintensive interventions directed to younger patients progressing to MRD positivity posttransplant are of interest for further evaluation, optimizing delivery will be challenging because of the variables of relapse kinetics and tolerability.
Comment on case presentation
A 62-year-old man with a normal white blood cell count was diagnosed with cytogenetically normal AML, and subsequent molecular studies for ELN 2017 adverse and good risk mutations, including mutated-NPM1, were negative. He was fit enough to be treated with 2 courses of induction chemotherapy (standard UK NCRI AML treatment). MRD response was assessed by flow cytometry. He achieved a CR but was MRD positive by flow cytometry (at 0.12%) after the second cycle. Because of deterioration in performance status, his comorbidities, and preference, further treatment options did not include transplant. He was known to have IDH1 R132 and DNMT3A mutations and was considered for novel regimens or azacitidine. Should he be monitored if he has further treatment and how?
Even if this patient were younger, he has a very high risk for relapse by his postinduction MRD status, even when treated with 3 or 4 courses of standard induction/consolidation chemotherapy (3-year CIR 89%, data from NCRI AML17 trial for younger adults5 ). Because he could not proceed to allogeneic transplant, other treatment options include azacitidine maintenance (although it is uncertain whether MRD-positive patients derive benefit) or, when available, novel regimens incorporating Bcl287 or IDH1 inhibition (venetoclax and ivosidenib, respectively).22 There are no recommendations available for MRD monitoring off-trial in the setting of less intensive treatment. Because he is MRD positive by flow cytometry, conversion to MRD negativity by this assay (observed in 32% of older AML patients attaining CR/CRi with venetoclax and low-dose cytarabine87 and in 40% treated by hypomethylating agents88 ) may be encouraging for a more durable response, although not as yet a surrogate for survival. Parallel monitoring of IDH1R132 mutation20-22 may be particularly informative for tracking on-target effect of an IDH1 inhibitor. Clearance of IDH1 mutations by digital PCR (limit of detection 2 to 4 × 10−4) in this context appears promising as a surrogate for outcome.22
Concluding remarks
AML MRD evaluation in clinical practice is happening and will continue to increase. Upfront intensification that includes allogeneic transplantation plus incorporation of available novel agents and maintenance schedules expand current possibilities to improve outcomes but require evidence from randomized clinical trials to establish benefit. MRD-guided therapy added to diagnostic genetic profiling has the potential to target these therapeutic options appropriately and, thus, improve the ratio of benefit to toxicity and costs. Although MRD at single time points has strong prognostic (and, for some treatments, predictive) value at a cohort level, consecutive measurements of currently recommended MRD targets during and after treatment are more likely to provide accurate information for individual patients by tracking any increase in their MRD levels. This also reduces the potential impact from false-negative and false-positive MRD results inherent in any single MRD test.
As in the case of acute lymphoblastic leukemia, chronic myeloid leukemia, chronic lymphocytic leukemia, and myeloma, trial participation is likely to continue as a key factor for enabling familiarity with the clinical use of MRD, as well as advancing methodology. Clinical trial networks provide resources and direction in addition to investigating unresolved questions. This has been observed, for example, in the United Kingdom; MRD-informed treatment of non–acute promyelocytic leukemia has been integrated into UK NCRI trials since 2012 with availability of coordinated advice for interpretation of results and management decisions. An important task for the next few years will be the evaluation of error-corrected NGS MRD assays, particularly in those AML subgroups without available sensitive PCR assays. Because there is no uniform marker of leukemia or clonality in AML, unlike chronic myeloid leukemia and lymphoid malignancies, the most appropriate prognostic MRD platform and whether orthogonal testing best measures leukemic reservoirs of relapse will depend on AML subtype. In this regard, AMLs with greater intratumoral genetic heterogeneity (and, consequently, also with increased mechanisms for treatment escape) are particularly challenging for tracking relevant MRD by mutations.
Acknowledgments
The authors thank Richard Dillon (UK NCRI Group) and Adriana Plesa and Christophe Roumier (coordinators of Flow AML MRD on behalf of the Acute Leukemia French Association (ALFA) laboratories and lead investigators) for the analyses and preparation of their contributed figures, as well as helpful discussions. They also thank Naeem Khan (University of Birmingham) for assistance with mass cytometry and gratefully acknowledge their ELN colleagues for major contributions to information in this review, including a continuing tribute to David Grimwade. S.D.F. and Richard Dillon thank Nigel Russell, Alan Burnett, Robert Hills, Charlie Craddock, Paresh Vyas, Brian Huntly, and other members of the NCRI AML Working Group for advice and enabling evaluation of MRD in the NCRI AML trials. S.D.F. also gratefully acknowledges present and past laboratory colleagues of the NCRI MRD network, including Kathleen Gallagher, Georgia Andrew, Nick McCarthy, Marlen Meztner, Edward Theakston, Adam Ivey, Jelena Jovanovic, Nicola Potter, and Amanda Gilkes.
This research was supported in part by the Intramural Research Program of the National Heart, Lung, and Blood Institute of the National Institutes of Health.
Correspondence
Sylvie D. Freeman, Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham B15 2TT, United Kingdom; e-mail: s.freeman@bham.ac.uk.
![(A) Suggested management algorithm for patients with AML with a molecular MRD target. [1] ELN favorable risk patients with <4-log reduction in NPM1-mutant transcripts after first induction are shown to benefit from a CR1 allograft,4 and any positivity in the peripheral blood (PB) after second induction is associated with a very high risk for relapse.3 [2] MRD positivity > 200 copies per 105ABL (ie, molecular persistence) and serially increasing transcript levels after treatment (ie, molecular progression) reliably predict relapse.7 [3] At the end of treatment, patients with CBF AML with high or serially increasing transcript levels are destined to relapse (relevant thresholds are >500 copies [per 105ABL] of RUNX1/RUNX1T1 in the BM or >100 copies in the PB, and > 50 copies of CBFB/MYH11 in the bone marrow [BM] or >10 copies in the PB).7 Salvage according to (C) should be considered for these patients, although there is no evidence that this improves outcome. Conversely, patients with low copy numbers below these thresholds can be safely monitored according to (B). [4] Although CBF patients with an early unfavorable MRD response have a higher risk for relapse, there is insufficient evidence to warrant treatment change7; however, this may prompt initiation of an early donor search. Salvage may be considered in cases with extremely poor early response or if there is an increase while on treatment (ie, molecular progression). [5] Although there is no evidence that standard-risk patients who remain MRD positive benefit from transplant, this is a reasonable approach and is adopted in the current NCRI AML19 and MyeChild01 protocols. (B) Suggested algorithm for sequential monitoring after treatment. Patients with conversion to MRD positivity, confirmed on a second sample with >1-log rise, should be diagnosed with molecular relapse and treated as shown.7 (C) Possible peri-transplant management strategy. [1] Patients with an NPM1 mutation without FLT3 ITD who have transcript levels below 1000 copies in the BM or 200 copies in the PB have a very good outcome after allograft, it is uncertain whether these patients benefit from salvage chemotherapy.89 [2] Patients with high levels of MRD after salvage, without an adequate response to donor lymphocyte infusion (DLI), as well as those to whom these standard therapies cannot be given should be considered for investigational approaches. Figure by Richard Dillon, NCRI Group.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2019/1/10.1182_hematology.2019000060/5/m_hem2019000060cf1.png?Expires=1770634789&Signature=3BHoXis2mcbcPz-zu4PGfoKVgof6lMvL-rcECuDYV1RXem-QXppWEmDw06aSAVCDSR3wYP~x-ZLWpASUnumiIl8oKSo8t-5ky4KfRn-zAs22YvGYD~KnHWxY6DXFMsKc1TRiYbe5uTmYDJF-n8bYC11yMCY4n5JG6oAwU0Bhr4DvsY17v2TyBp~9g5BgkxPZ6F9cmeJ0RCCKPpSJ456swBUzIypcMa7Ac7z1qUKQHbbVC3J8fFEkxdsvSZpx5id5oA4p84VBuZi9P4vz8YkRwlo2Lo1Pk5wmV-P7pWrxm5~k9rhsUJa33~HsL0N2r8QtaisosuCUjWGRG1ds5yRJdQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![(A) Strategy applied for flow cytometric AML MRD multicenter harmonization by the ALFA Intergroup. [1] Rationale of AML MRD flow panel design was based on simplicity, reproducibility, and cost. Tube 1 was a core combination for LAIP detected at diagnosis and/or by different-from-normal analysis, tube 2 was targeted to aberrancies of CD34+CD38− cells (immunophenotypic LSCs), and tube 3 was an optional development tube for monocytic aberrancies. [2] Flow cytometer fluorescent settings were harmonized (“mirrored”) between Canto vs Navios cytometer platforms. Voltages were set to reach target mean fluorescence intensity (MFI) values by acquisition of rainbow calibration beads without compensation for fluorescent channels FL1 to FL8 on the Canto cytometers; these rainbow bead settings were transposed to Navios cytometers by applying MFI target = Canto target/256. Mirrored (superimposable) target peaks for both cytometers are shown for FL1 and FL8 fluorescent channels with an example of resulting comparable antibody profiles between cytometer platforms. (B) [3] Quality controls for reproducibility of staining profiles from harmonized cytometer settings/sample processing between cytometers/laboratories. Examples shown are for CD117 and CD38 expression intensity on CD34+ gated mononuclear cells of tube 1 from 10 shared BM samples stained and then acquired on Canto or Navios flow cytometers. Intensity profiles are similar between cytometer platforms for each sample. [4] External quality assessment for all harmonized steps from preanalytical to final gating analyses by distribution of a normal BM to 22 participating laboratories (cytometer platforms: 12 Cantos, 10 Navios). Example shows that strong reproducibility can be achieved in the detection of rare events (shown for CD34+CD38−) among 22 participating laboratories. Figure by Christophe Roumier and Adriana Plesa, ALFA.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2019/1/10.1182_hematology.2019000060/5/m_hem2019000060cf3.png?Expires=1770634789&Signature=47AaM~4BMTGiNNWm32490G8qpn79pJReQ-WY15SqBBFbccTxYMLZpsTtpTJMp-nZS2vyDuyJ7xtz76D78cc7PjfDssMxhudzKh7rkm902P6j3XhOlF5-EMoeWaDqCFvl~yZVV-uuskZr9xELx5RQ0mXq-Ct25Y7rU2C9Sn2dLV71Ljmb~LYY83sikrpvD1A~MdBKJFMSvYMx30m0umYXXt0h4IfthuaxaEKdQXfRovLD4D891Jbgmz0CwGkZvTvoVQENCXAb5SJ5-ETS-GTFNbrHGdJjVunIihETfy4u3TKNkS9J33FYl~beCNhGjb7moZDDuEnFD3Iq9ly34DAebg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)