Abstract
With access to safe factor products, the life expectancy of persons with hemophilia (PWHs) has increased almost 10-fold over the past 7 decades. Unfortunately, hand in hand with this success comes the burden of aging. As PWHs age, they are subject to develop many of the same risk factors as the general population, including increasing rates of hypertension, obesity, and diabetes. Such comorbidities predispose them to chronic diseases, such as cardiovascular disease and chronic kidney disease, although how their coagulopathy affects the expression of these conditions remains unclear. The older hemophilia population faces additional challenges, such as chronic joint arthropathy, which provokes falls and fractures, and complications related to HIV and hepatitis C infections, which greatly affect the incidence of cancer and liver disease. In light of the paucity of evidence-based guidelines to direct therapy, a new challenge has arisen for hematologists to optimally manage these complex age-related issues. In general, elderly PWHs should be treated similarly to their peers without hemophilia, with the addition of factor replacement therapy as appropriate. Primary prevention of risk factors should be emphasized, and close coordination between specialties is essential. This review will focus on common complications affecting the older hemophilia population, including cardiovascular disease, malignancy, liver disease, renal insufficiency, and joint disease.
Learning Objectives
To describe common complications affecting the older hemophilia population, including cardiovascular disease, malignancy, liver disease, renal insufficiency, and joint disease
To understand special precautions that need to be taken for the management of these medical comorbidities in the hemophilia population
Hemophilia is the most common X-linked heritable disease. It results in low levels of factor VIII (hemophilia A) or factor IX (hemophilia B) and affects ∼400 000 people worldwide.1 For persons with hemophilia (PWHs), the life expectancy in industrialized nations has increased staggeringly from 7.8 years in 1939 to >70 years in 2001, mainly because of the availability of effective and safe factor concentrate.2 The contamination of plasma-derived factor concentrates with HIV from 1978 to 1986 and hepatitis C virus (HCV) before 1992 devastated the hemophilia community.2 Currently, patients with severe hemophilia unaffected by HIV have a median survival in the 6th or 7th decade of life; with the use of highly active antiretroviral therapy, the survival of PWHs infected with HIV has also improved significantly since the 1990s, with 27%-39% surviving 20-25 years after seroconversion.3-5 With modern day safe treatment, we now happily face a growing population of PWHs with advancing age (Figure 1). However, there is relatively little experience in treating this older population and their developing age-related diseases, raising a new challenge for hematologists. The focus of this review is on hemophilia and age-related complications, including cardiovascular disease (CVD), malignancy, liver disease, renal insufficiency, and joint disease.

Comparison of age distribution of persons with all severities of hemophilia A from 2005 to 2014.
Comparison of age distribution of persons with all severities of hemophilia A from 2005 to 2014.
CVD
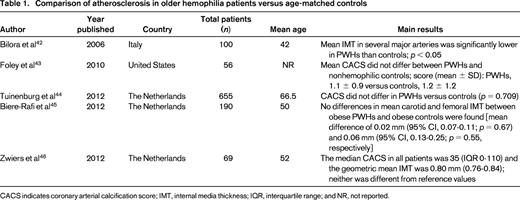
As the hemophilia population ages, they are subject to the same risk factors to develop atherosclerotic disease as the general population, including hyperlipidemia, smoking, obesity, metabolic syndrome, and especially hypertension (HTN) and chronic kidney disease (CKD).6,7 HIV may also play an important role in the development of CVD in older PWHs. Infection with HIV is associated with an increased incidence of vascular disease, the mechanism of which is complex and beyond the scope of this review.8 The majority of available studies report a resultant similar amount of atherosclerosis in PWHs compared with controls (Table 1). However, whether hemophilia exerts a biologically protective effect through lower FVIII and IX levels on the actual incidence of thrombotic events in CVD remains unclear.
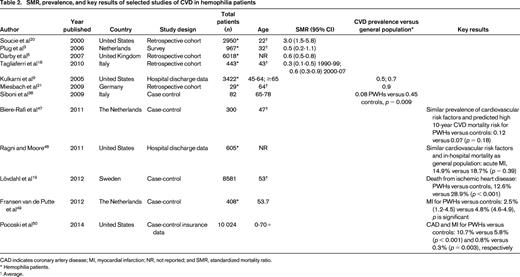
In 2005, Kulkarni et al9 retrospectively evaluated a cohort of 3422 males with hemophilia in the United States and found the prevalence of ischemic heart disease (IHD) to be 0.05% in those aged <30 years and 15.2% in those aged >60 years. The authors concluded that, although less common than the general population, older hemophilic patients remain at risk of IHD. Controversially, some analogous studies report a protective effect of hemophilia on the incidence of CVD, whereas others show an increased cardiovascular risk profile. The studies are heterogeneous and difficult to directly compare because of disparate age ranges and outcomes (see selected studies in Table 2). Biere-Rafi et al10 conducted a systematic review of arterial disease in hemophilia in 2010 and found nonsignificantly reduced cardiovascular mortality in PWHs compared with healthy controls [standardized mortality ratio (SMR), 0.51; 95% confidence interval (CI), 0.24-1.09]. Given conflicting reports in the literature, there is clearly a need for a large prospective study to determine the risk of CVD in hemophilia patients. Preliminary results from an ongoing U.S. cross-sectional analysis of CVD (CVD in Hemophilia Study) found that in 148 men aged 54-73 years with moderate or severe hemophilia, older men with hemophilia have high rates of risk factors for CVD, including HTN (64.2%), dyslipidemia (35.1%), and renal insufficiency (29.3%). However, the prevalence of reported CVD was low at 10.1%, suggesting that men with hemophilia may be protected from forming pathogenic thrombi. A formal comparison with age-matched controls is planned once enrollment is complete.11
Currently, great treatment uncertainties exist in the management of PWH with IHD, given the risk of bleeding with antiplatelet and anticoagulation agents in these patients. There is an urgent need for data to not only define the safety of antiplatelet agents but also the recommended dose of factor replacement, because evidence-based treatment guidelines are lacking. Proposed recommendations for treating hemophilia patients with CVD generally parallel those in individuals without hemophilia, with the addition of adequate factor replacement.12,13 For primary prevention of CVD and stable angina, upfront medical management of cardiovascular risk factors is key. Decisions regarding the risk of long-term antiplatelet agents need to be made on an individual basis. In patients with mild disease (ie, factor levels >5%), long-term low-dose aspirin (81-100 mg) may be considered safe, but patients with severe hemophilia or inhibitors would require factor prophylaxis. A similar risk–benefit analysis must be performed when considering anticoagulation for atrial fibrillation, and there may be a lower threshold to proceed to direct cardioversion.14 Long-term anticoagulation is relatively contraindicated in severe hemophilia, but if deemed necessary, one would need to carefully consider the half-life and reversibility of the chosen anticoagulant.
Factor replacement should be used for the treatment of acute coronary syndromes when anticoagulant and antiplatelet medications are used, with recommended factor levels of at least 30%-60%.15 In this regard, continuous factor infusion rather than bolus administration has been proposed to avoid extreme peaks and trough levels.16 The Dutch guidelines recommend peak factor levels of 80% for 48 hours when arterial access is required for primary coronary intervention with platelet inhibition; they also view hemophilia as a contraindication to systemic fibrinolysis, although this has been debated in mild disease.12 Radial artery access has been suggested widely over femoral access given the lower risk of bleeding complications with this approach. If coronary stenting is required, bare metal stents are preferred over drug-eluting stents because of limited duration of required dual antiplatelet therapy.
Treatment options for those individuals who require cardiac surgery has mainly only been addressed in case reports.13 Procedures requiring a coronary bypass pump with systemic anticoagulation are clearly high risk, and off-pump or more minimally invasive operative options are favored. Bioprosthetic valves are preferred over mechanical prosthesis to limit the required duration of anticoagulation. The use of antiplatelet and anticoagulant agents in the setting of cardiac disease requires rigorous monitoring for bleeding complications and a close relationship between treating disciplines is paramount. Prospective validation for the above treatment recommendations are currently in need.
Cancer
The risk of developing cancer increases with age, and the same holds true in the aging hemophilia population. Moreover, cancer risk is influenced strongly by viral status. According to a recent report published by the American Thrombosis and Hemostasis Network, the current estimate of HIV infection in those with FVIII deficiency is ∼21%, and HCV is slightly >50% in those aged ≥30 years.17 As in the general population, hemophilia patients infected with HCV have a greater risk of developing hepatocellular carcinoma (HCC), and HIV infection increases the risk of malignancies, such as non-Hodgkin lymphoma, basal cell carcinoma, and Kaposi sarcoma, among others.
Given the available data, it is not clear whether hemophilia patients have a higher incidence of cancer compared with age-matched controls. Some population-based studies report lower mortality rates from non-HCC cancers in PWHs compared with the general population,5,18 whereas others found malignancy to be a leading cause of death in PWHs.19,20 In 2009, Miesbach et al21 reported that the prevalence of cancer was 5 times higher in hemophilia patients aged ≥60 years compared with aged-matched controls (28% versus 5.2%), with a 4-fold increase in the prevalence of cancers in PWHs other than HCC, including colon, prostate, skin, bladder, and lymphoma. Huang et al22 similarly reported a higher rate of cancer in PWHs compared with the general population [relative risk (RR) 1.66; 95% CI, 1.06-2.59] in Taiwan, even after excluding viral-related malignancies. In contrast, in a recent review, Franchini et al23 concluded that HCV- and HIV-associated cancers are more common in the hemophilia population in those affected by the virus(es) but did not find evidence of an increased incidence of other malignancies when compared with the general population.
Similar to the general population, age appropriate cancer screening is indicated in those with hemophilia. Given the increased risk of bleeding, special care needs to be taken when biopsies are pursued and during treatment with chemotherapy or radiation because such therapies can induce thrombocytopenia or cause mucosal injury that can predispose to hemorrhage. Those infected with HCV who develop HCC are in need of particular consideration because the resultant liver dysfunction can lead to additional coagulopathy and thrombocytopenia. When diagnosing and treating malignancy, it is crucial to provide adequate factor replacement to minimize the risk of bleeding. Although limited by a lack of evidence, Franchini et al23 suggest keeping factor levels >50% on the day of biopsy as well as 3-4 days after. For more invasive surgical procedures, the recommended factor level is >80% for the first 3 days, followed by >50% for 7-15 days after the procedure; neurosurgical procedures may require higher target levels. Use of pharmacological venous thromboembolism prophylaxis after oncological surgery remains controversial in PWHs.
With regard to treatment, antineoplastic treatments should mimic what is recommended for the general population. Some authors have recommended initiating prophylaxis when treatment is complicated by severe thrombocytopenia (platelet count <30 × 109/L), and, in some patients, platelet transfusion should be considered.14 It is also prudent to be mindful about potential drug side effects on the hemostatic system. For example, there are several vascular endothelial growth factor inhibitors on the market that can exacerbate bleeding tendency, and alternative agents should be selected if possible in PWHs.
Liver disease
The leading cause of liver disease in PWHs is infection with HCV. Most chronic HCV infections result in liver inflammation and fibrosis, and ∼20-30% of affected patients progress to cirrhosis.24 The risk of cirrhosis increases with time after initial infection, advancing age, and coinfection with HIV. In addition to a decrease in anabolic liver function leading to less production of coagulation proteins, bleeding manifestations can be exacerbated by the thrombocytopenia and varices common in cirrhotic patients. It is exciting that a phase 2b trial for HCV treatment in PWHs and other inherited bleeding disorders with novel interferon-free regimens (ledipasvir/sofosbuvir, sofosbuvir/ribavirin) has been completed recently (results pending). Novel agents against HCV in PWHs may be much more tolerable and thus greatly influence the natural history of liver disease in this population. Orthotopic liver transplantation is another treatment option; 10% of all liver transplants are performed on PWHs infected with HIV/HCV, although shortened pretransplant survival compared with the general population has been seen.24
As mentioned previously, HCC is common in the aging hemophilia population because of a high prevalence of risk factors, including HCV infection, cirrhosis, and HIV coinfection. Given the high frequency of HCV infection in elderly PWHs, it is important to note the American Association for the Study of Liver Diseases guidelines for HCC screening were updated in 2011: surveillance for HCC should be pursued in HCV-positive patients with cirrhosis by liver ultrasound every 6 months, and diagnosis of HCC should be made by additional imaging and/or biopsy. Serum α-fetoprotein testing has fallen out of the guideline given its inadequate specificity and sensitivity.25 In light of advances in the treatment and eradication of HCV, a recent meta-analysis of observational studies by Morgan et al26 evaluated the effect of HCV eradication on the development of HCC. They found that, in persons with sustained virologic response to treatment, the RR of developing HCC was 0.24 (95% CI, 0.18-0.31). It is important to note that these patients still require HCC screening.
CKD
CKD is a common condition with increasing prevalence in the United States. The National Health and Nutrition Examination Survey found that stage 3-4 CKD increased from 0.2% in 1988-1994 to 0.7% in 1999-2004 in those aged 20-39 years; a similar rise was seen in those aged ≥70 years from 27.8% to 37.8%.27 These data also reflect the marked association between CKD and advancing age.
PWHs appear to have a higher incidence of CKD compared with the general population. Kulkarni et al28 reported a higher rate of CKD in men with hemophilia (4.7 in 1000 patients per year versus 2.9 in 1000 in age-matched controls) in a study that analyzed hospital discharge data in 3422 men with hemophilia in 6 U.S. states. Risk factors for CKD included HTN, HIV infection, older age, non-white race, or a history of renal bleeds.28 In addition, PWHs may have higher mortality because of their renal disease. Soucie et al20 found that U.S. men with hemophilia were 50 times more likely to die from renal disease compared with the general population, confirming an older study by Rosendaal et al.29 Preliminary analysis of 148 older men with moderate-to-severe hemophilia in the ongoing U.S. CVD in Hemophilia Study found an increased incidence of renal insufficiency: 29.3% of the cohort has an abnormal creatinine clearance, markedly worse than expected in the general U.S. population. Interestingly, in this study, subjects with renal insufficiency did not appear to have higher rates of DM, HTN, or HIV, suggesting that the pathogenesis of CKD in hemophilia may be different from the general population.11 This study has been extended to better determine the prevalence of CKD in the elderly hemophilia population.
Potential etiologies for the increase in CKD include the higher rate of HTN seen in hemophilic males, as well as urinary tract bleeding (the most common hemorrhagic site after hemarthrosis).30 These chronic insults may result in structural kidney damage, although the latter association remains speculative. Hemophilia treatments may induce injury, including urinary obstruction from the use of antifibrinolytic therapy31 and nephrotic syndrome in patients with FIX inhibitors undergoing immune tolerance therapy. There is a clear association between viral infections common in the aging hemophilia population (HIV and HCV) and well-documented renal complications, such as HIV associated nephropathy, viral-induced glomerulonephritis, interstitial nephritis, and membranous nephropathy.32 One of the most effective treatments for viral-related CKD is antiviral therapy; unfortunately, medications used to treat these viral infections can also be nephrotoxic. Data on hematuria and its relationship to renal disease are conflicting, and the role of renal bleeding and its relationship to CKD requires additional study.33-35
The management of end-stage renal disease (ESRD) in the hemophilia population is complex, because minimal data are available to guide decision making. In those who need renal replacement therapy, the decision between peritoneal dialysis (PD) versus hemodialysis (HD) needs to incorporate patient-specific factors. HD exposes patients to increased risk of bleeding, and, if heparin is used to prevent the extracorporeal circuit from clotting, standard prophylactic factor replacement is recommended with each dialysis session (ie, 25-40 IU/kg FVIII 3 times per week).32 Alternatively, HD can be performed without the use of heparin. The risk of major bleeding with HD makes PD more attractive, given that specific factor replacement is not required with PD. However, PD may not be the best approach for someone with cirrhosis and ascites given the increased risk of infection.
If HD is chosen as the treatment modality, central venous access is mandatory. Before placement of a temporary vascular device, it is recommended that the factor level should be 100% and then kept between 50 and 100% for 3 days after the procedure with a goal of 30%-75% for an additional 1-5 days.36 Ports are preferred because of longevity and lower infection rates. A similar regimen is recommended for permanent arterial venous fistula placement with daily infusions extending an additional 5-6 days.32 Renal transplant may also be a suitable option for patients with ESRD, and successful outcomes have been reported in the literature for PWHs. Current guidelines generally recommend preoperative factor levels of 80%-100%, >50% for 1 week after the procedure and >30% for the late postoperative period.32
Given the rising age of the hemophilia population, a growing number of patients will develop renal disease in the near future. Hematologists need to be aware of this potential problem and control reversible risk factors to prevent additional damage. Both prevention and proper management of existing renal disease takes coordination among numerous subspecialty physicians, and early nephrology involvement is crucial.
Joint disease
Arthropathy is the most common challenging condition facing hematologists who care for the aging hemophilia population. In most countries, PWHs who were diagnosed before 1987 were not treated routinely with prophylactic factor replacement during their childhood, because this practice was not yet standard of care given the fear of viral contamination.37 As a result of chronic bleeding into target joint spaces, the older hemophilia population is living with impaired range of motion, joint instability, contractures, muscular atrophy, and chronic synovitis. These mechanical problems inevitably lead to impaired mobility, a risk factor for obesity, which in turn can put additional strain on damaged joints. In an Italian study of men aged ≥65 years with severe hemophilia, almost all patients had arthropathy with a significantly higher pain score compared with age-matched controls. In addition, PWHs had an increased risk of falls, functional debility, and a 7-fold higher rate of joint replacement surgery.38
It is important to recognize hemophilic arthropathy to manage chronic pain and mitigate confounding factors that could lead to falls or fractures. Gay et al39 recently published a retrospective analysis of 382 PWHs and reported a staggeringly increased risk of fracture in the hemophilia population compared with the control population (RR, 10.7; 95% CI, 8.2-14.1; p < 0.0001). Fracture risk correlated with both disease severity and age: PWHs aged >31 years were twice as likely to suffer fracture compared with the younger population (RR, 2.15; 95% CI, 1.26-3.65; p = 0.0047).39 The heightened risk of fractures is postulated to be secondary to decreased bone mineral density from chronic inflammation and decreased weight bearing on hemarthrotic joints.
Given the increased rate of joint morbidity, efforts are needed to develop preventative strategies. Secondary prophylaxis generally leads to a reduction in bleeding, but its efficacy in improvement of orthopedic function remains unclear.37 Additional studies to explore the optimal use of prophylaxis in older individuals are needed. A recent review by Forsyth et al40 discussed the role of exercise in hemophilic arthropathy. The authors promote a regular exercise program incorporating strength training, aerobics, balance, and flexibility in PWHs to improve joint function, stability, and quality of life. In addition, such exercises can increase bone mineral density, protecting PWHs from osteoporosis and resultant pathological fractures further contributing to decreased mobility. Such programs are ideally conducted under the supervision of a trained physiotherapist familiar with hemophilia. Consideration should be given to prophylactic factor replacement before exercise in those with severe hemophilia.40
Joint replacement surgery is becoming more common in the older hemophilia population, given the combined risk factors of age and recurrent hemarthrosis in causing debilitating arthritis. The most common target joints are the knees, ankles, and elbows. Of these sites, knee arthropathy is associated with the most morbidity, and total knee replacement is the most common joint replacement performed in PWHs. The indication for joint replacement is advanced arthritic disease that is affecting function and quality of life. This procedure is best performed at a hospital associated with a hemophilia treatment center where orthopedic surgeons, nurses, social workers, physical therapists, and hematologists are familiar with this patient population. Adequate factor replacement perioperatively is paramount and needs to be monitored carefully by a hematologist experienced with treating hemophilia. The role of pharmacological venous thromboembolism prophylaxis in PWH after hip or knee replacement remains controversial. Early ambulation should be encouraged, with pharmacologic prophylaxis considered on a case-by-case basis.41
Conclusion
Given recent treatment progress in the management of hemophilia since the 1970s, hematologists are seeing more and more hemophilia patients with advancing age. Comorbidities that trend with age, such as high cholesterol, HTN, and obesity, are now affecting the older hemophilia population. Such issues predispose them to chronic diseases, such as CVD and CKD, although how their coagulopathy affects these conditions is still essentially unknown. The older hemophilia population faces additional complications, such as joint arthropathy and complications related to HIV and HCV infection, which affect the incidence of cancer and liver disease in this population. There is still a lack of evidence to base guidelines on how to treat the older hemophilia population and age-related comorbidities, and additional prospective studies and experience are needed for us to learn how to give optimal care to this special population.
Correspondence
Suman L. Sood, Hemophilia and Coagulation Disorders Program, MIB C351A, University of Michigan, Ann Arbor, MI 48109-5848; Phone: 734-615-4762; e-mail: sumisood@med.umich.edu.