
Most disease entities in hematology have a long and storied history, being described decades ago and undergoing progressive refinement as increasingly sophisticated diagnostic tools are developed. Chronic myeloid leukemia is a typical example, first described in 1845 by J.H. Bennett and colleagues as “suppuration of the blood” and now understood as a manifestation of BCR-ABL1 rearrangement in a hematopoietic stem cell, with highly effective therapies that target the underlying genetic lesion. Most recent research in the diagnosis and classification of hematologic neoplasms has focused on better defining existing disease entities, often through deep genetic interrogation, or improving their risk-stratification. An exciting exception to this paradigm is the recent discovery of VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome (Figure).
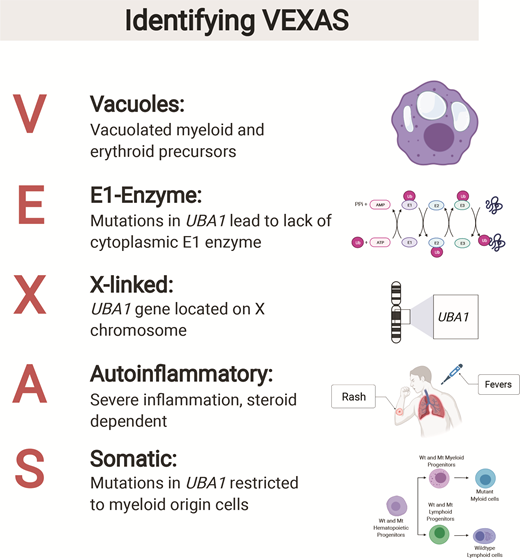
Figure created by and used with permission from Daniela Cardona, National Institutes of Health.
Figure created by and used with permission from Daniela Cardona, National Institutes of Health.
VEXAS syndrome is an adult-onset disease characterized by somatic mutation of the UBA1 gene in hematopoietic progenitor cells and manifesting clinically as macrocytic anemia and autoinflammation.1 UBA1 encodes the major E1 enzyme that initiates ubiquitylation and is located on the X chromosome; this X-linked gene does not undergo X-inactivation in female patients and thus, its mutational inactivation results in the disease almost exclusively affecting male patients. VEXAS was initially reported in a series of 25 men by Dr. David Beck and colleagues after broad exome sequencing had revealed somatic UBA1 mutations in an initial cohort of three men with anemia and systemic inflammatory disorders.2 All patients had macrocytic anemia and various inflammatory disorders including relapsing polychondritis, Sweet syndrome, and vasculitides; most patients were thrombocytopenic. All patients who had undergone bone marrow biopsies showed prominent vacuolization of the bone marrow early in myeloid and erythroid precursors. While vacuolization of hematopoietic elements is a nonspecific finding that can be seen in numerous reactive conditions such as copper deficiency as well as in myeloid neoplasms,3 the ubiquitous association of this finding with UBA1 mutation and systemic inflammatory disorders is striking.
Dr. James Poulter and colleagues4 sequenced the UBA1 gene in 18 male patients older than 40 years with the combination of severe systemic inflammatory disorders, cytopenias, and bone marrow dysplasia (including vacuolization). They found UBA1 mutations in 10 patients, including two with novel mutations different from the p.Met41 substitutions found in the original report.2 In a subset of cases in the series from Dr. Beck and colleagues and Dr. Poulter and colleagues, the dysplastic bone marrow features were consistent with a diagnosis of myelodysplastic syndrome (MDS). Subsequently, Dr. Ifeyinwa Emmanuela Obiorah and colleagues5 studied the detailed hematologic and bone marrow features of 16 patients with VEXAS syndrome and found MDS (based on significant morphologic dysplasia) in 10 patients; all had low blast counts and none progressed to acute leukemia. In some patients with MDS, the UBA1 mutation was accompanied by additional somatic mutations in genes implicated in MDS, such as DNMT3A, or abnormal findings on karyotype. An additional association with VEXAS syndrome is the occurrence of plasma cell neoplasms in 20 percent of patients observed in the original report by Dr. Beck and colleagues, as well as some cases of monoclonal B-cell lymphocytosis in the article by Dr. Obiorah and colleagues; since the UBA1 mutation appears to be restricted to myeloid cells in the blood, this association with clonal plasma-cell or B-cell proliferations remains enigmatic.
These recent studies have revealed a hitherto unknown disease caused by a single somatically acquired gene mutation in a key cellular enzyme and resulting in a unique combination of clinical sequelae. Although bearing some similarities to MDS (clonal mutated hematopoiesis, macrocytic anemia due to ineffective erythropoiesis, and morphologic abnormalities of hematopoietic cells), the hematologic features of VEXAS are unusual: The anemia tends to be refractory to hypomethylating agents or erythroid-stimulating agents, yet progression to excess blasts is rare and progression to acute myeloid leukemia has not yet been reported.6 The identification of VEXAS has further crystallized the association of clonal hematopoiesis to systemic inflammation, which has been previously reported in some overt myeloid neoplasms such as chronic myelomonocytic leukemia7 as well as in clonal hematopoiesis of indeterminate potential.8 Better understanding of the biology of this association could lead to novel therapies for both myeloid neoplasms and rheumatologic diseases, and VEXAS serves as a paradigm of these “hematoinflammatory diseases.”1 More widespread awareness of VEXAS is important in order to identify affected patients and investigate optimal therapeutic approaches, as this syndrome is associated with significant morbidity and mortality due to transfusion dependency in many patients and the often severe rheumatologic manifestations.
Vast advances in comprehensive sequencing have undoubtedly improved our understanding of the genetic basis of hematologic diseases, led to more objective criteria for their diagnosis, and stimulated the development of effective targeted therapies. However, lest we fall into the trap of thinking we have a full understanding of the landscape of hematopoietic neoplasms, VEXAS reminds us that we still have much to learn. The work of Dr. Beck, Dr. Poulter, and Dr. Obiorah and their colleagues, as well as others who brought VEXAS to light in 2021, teaches those of us who study blood disorders to keep looking deeper. A new entity may lie just around the corner!
Competing Interests
Dr. Hasserjian indicated no relevant conflicts of interest.
