

Abstract
Acquired hemophilia is a rare bleeding disorder that predominantly affects older people with potential underlying comorbidities, including cardiovascular and thrombotic risk factors. The current standard therapies with hemostatic agents for acute bleeding and immunosuppression often require inpatient management, are not approved for routine bleeding prophylaxis, and contribute to the high mortality in this population. Emicizumab is a factor VIII (FVIII) mimetic approved for bleeding prophylaxis in congenital hemophilia A with and without FVIII inhibitors. Given subcutaneously, it may allow easier outpatient bleeding prophylaxis and reduce intensity of immunosuppression. This article summarizes the currently available data on the efficacy and safety of emicizumab in acquired hemophilia A.
Learning Objectives
Recognize the current challenges of current treatment of acquired hemophilia A
Summarize the current experience with emicizumab in AHA
Describe the current safety profile of emicizumab use in AHA
CLINICAL CASE 1
One month after the birth of her second child, a 34-year-old gravida 2, para 2 woman noticed easy bruising and swelling of her left arm and leg that slowly resolved (Figure 1. A/B). Approximately 10 months postpartum, she reported to the emergency department with acute onset right arm swelling with severe pain and numbness (Figure 1. C). She underwent emergent fasciotomy for compartment syndrome, which was complicated by severe intraoperative bleeding that required 14 units of red blood cells (RBCs) over the next week. She was subsequently diagnosed with acquired hemophilia A with a factor VIII (FVIII) activity of 6 IU/dL and a FVIII inhibitor of 11 Bethesda units (BU). Treatment with activated prothrombin complex concentrate (aPCC) was initiated with significant improvement in bleeding, but she continued to saturate her bandages every 2 to 4 hours and continued to require RBC transfusions. Her bleeding improved after switching hemostatic treatment from aPCC to recombinant porcine FVIII concentrate. However, 6 days after switching, her bleeding increased again and a porcine FVIII inhibitor measured 22 BU.
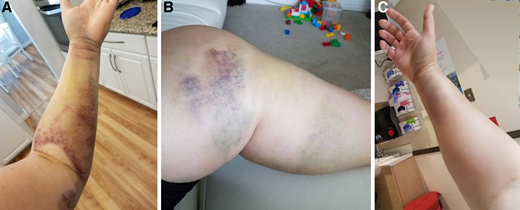
Bleeding in a case of postpartum-associated AHA. (A) Soft tissue bleeding in left arm; (B) soft tissue bleeding in left leg; (C) soft tissue bleeding resulting in compartment syndrome in right forearm.
Bleeding in a case of postpartum-associated AHA. (A) Soft tissue bleeding in left arm; (B) soft tissue bleeding in left leg; (C) soft tissue bleeding resulting in compartment syndrome in right forearm.
Introduction to acquired hemophilia A (AHA) and the challenges of currently available therapy
AHA is a rare autoimmune condition caused by an IgG autoantibody directed toward endogenous FVIII. This condition is idiopathic in about half of all cases but can be associated with other autoimmune diseases, malignancies, pregnancy, and certain drugs (particularly alemtuzumab, clopidogrel, and omalizumab).1 There also have been several case reports suggesting a potential association with COVID vaccination.2,3
Bleeding pattern and severity in AHA
Most patients present with spontaneous bleeding that is cutaneous or intramuscular or with retroperitoneal bleeding, but bleeding can also occur with trauma or procedures.4 Many bleeds are severe at presentation5 with significant drop in hemoglobin (Hb) necessitating blood transfusions, and they can quickly become limb-, organ-, or life-threatening.
Efficacy and limitation of currently available hemostatic therapies for AHA
Recombinant activated factor VII (rFVIIa) and aPCC are frequently used for acute hemostatic management of AHA (Table 1). These bypassing hemostatic agents were developed for patients with congenital hemophilia A and inhibitors. Due to their short half-lives and intravenous route of administration, both rFVIIa and aPCC often require hospitalization. Neither is approved for hemostatic prophylaxis outside the perioperative setting, and hemostatic under- or overcorrection cannot be monitored by a laboratory assay. Both contain activated factor VII, contributing to a potential risk of unwanted thrombosis.
Current hemostatic agents recommended for AHA
| Recombinant factor VII activated (rFVIIa) | Activated prothrombin complex concentrate (aPCC) | Recombinant porcine factor VIII (rpFVIII) | |
|---|---|---|---|
| Eptacog alfa, NovoSeven®RT | Anti-inhibitor coagulant complex, FEIBA® | Antihemophilic factor (recombinant), porcine sequence, Obizur® | |
| Indication | • Treatment of bleeding episodes and perioperative management in adults with acquired hemophilia30 | • Hemophilia A and B patients with inhibitors31 | • On-demand treatment and control of bleeding episodes in adults with acquired hemophilia A32 |
| Dosing | • 70-90 mcg/kg IV every 2-3 hours until hemostasis achieved30 | • 50-100 U/kg IV31 every 6-12 hours | • Initial dose 200 U/kg IV then titrate based on Factor VIII (FVIII) levels32 |
| Limitation of use | • Serious arterial and venous thrombotic events following administration have been reported30 | • Thromboembolic events reported during postmarketing surveillance, particularly following the administration of high doses and/or in patients with thrombotic risk factors31 • Contraindicated in acute thrombosis or embolism (including myocardial infarction)31 | • Safety and efficacy has not been established in patients with a baseline anti-porcine factor VIII inhibitor titer >20 BU32 |
| Recombinant factor VII activated (rFVIIa) | Activated prothrombin complex concentrate (aPCC) | Recombinant porcine factor VIII (rpFVIII) | |
|---|---|---|---|
| Eptacog alfa, NovoSeven®RT | Anti-inhibitor coagulant complex, FEIBA® | Antihemophilic factor (recombinant), porcine sequence, Obizur® | |
| Indication | • Treatment of bleeding episodes and perioperative management in adults with acquired hemophilia30 | • Hemophilia A and B patients with inhibitors31 | • On-demand treatment and control of bleeding episodes in adults with acquired hemophilia A32 |
| Dosing | • 70-90 mcg/kg IV every 2-3 hours until hemostasis achieved30 | • 50-100 U/kg IV31 every 6-12 hours | • Initial dose 200 U/kg IV then titrate based on Factor VIII (FVIII) levels32 |
| Limitation of use | • Serious arterial and venous thrombotic events following administration have been reported30 | • Thromboembolic events reported during postmarketing surveillance, particularly following the administration of high doses and/or in patients with thrombotic risk factors31 • Contraindicated in acute thrombosis or embolism (including myocardial infarction)31 | • Safety and efficacy has not been established in patients with a baseline anti-porcine factor VIII inhibitor titer >20 BU32 |
Recombinant porcine FVIII (rpFVIII, Obizur) is approved for both on-demand hemostatic treatment and prophylaxis in AHA. However, some patients present with anti-FVIII autoantibodies that cross-react with rpFVIII and reduce efficacy. Patients with AHA whose autoantibodies do not cross-react at presentation will often develop a de novo antibody against rpFVIII after ongoing exposure (typically 8 to 85 days after starting rpFVIII).6 This unfortunately makes rpFVIII efficacious in only a proportion of patients with AHA and for a limited duration. Waning efficacy should be closely monitored with FVIII activity levels. Once rpFVIII is no longer effective, patients with AHA must be restarted on bypassing agents for ongoing or recurrent bleeding.
The current international guidelines for AHA recommend hemostatic treatment with rFVIIa, aPCC, or rpFVIII rather than human FVIII concentrate for clinically significant bleeding and/or invasive procedures or surgeries.7 The efficacy of human FVIII is reduced by the anti-FVIII autoantibodies in patients with AHA. In a registry of 501 patients with AHA, FVIII products had a 70.1% rate of controlling frontline bleeds compared with 91.8% for the bypassing agents.8 Desmopressin (DDAVP) can be used to release endogenous FVIII stores but has limited utility in the presence of an autoantibody against FVIII.
The rebleeding risk after initial hemostasis and need for immunosuppression therapy
Approximately 50% of patients experience recurrent bleeding as per the prospective GTH-AH 01/2010 study of 102 subjects with AHA.9 The bleeding risk is particularly high in patients with FVIII activity of <20 IU/dL. Due to the high risk of recurrent bleeding and rarity of spontaneous remissions, immunosuppression is standard of care for patients with AHA.7 Frontline combination immunosuppression has historically been favored to try to irradicate the inhibitor as quickly as possible given the difficulties in treating bleeds with the available hemostatic agents, especially in the outpatient setting. Treatment recommendations for first-line immunosuppression (IST) traditionally have been glucocorticoids with cyclophosphamide or rituximab depending on the severity of the inhibitor and FVIII level.7,10 Adverse events from immunosuppression, however, are common, with a high mortality in those with secondary infections (30% of adverse events in the GTH/AH study were probably or definitely related to IST and 54% of those with infections died).11 Improved outpatient-based bleeding prophylaxis could reduce the intensity of immunosuppression and the risks of adverse events.
CLINICAL CASE 1 (continued)
After 3 weeks in the hospital, the patient achieved good temporary hemostasis with rpFVIII concentrate. Her fasciotomy was closed surgically and she did not require further RBC transfusion. She started on prednisone and cyclophosphamide, but her FVIII inhibitor titer remained detectable at 11 BU with an FVIII activity of 8 IU/dL. She developed a detectable anti-porcine FVIII inhibitor, and the efficacy of rpFVIII was waning. She remained at high risk of rebleeding but had a strong desire to return home to her 2 young children. To provide an outpatient hemostatic prophylaxis, the off-label use of emicizumab was started.
Efficacy of emicizumab for bleeding treatment and prophylaxis
Emicizumab is a bispecific antibody that binds activated factor IX and factor X mimicking the function of FVIII. Currently, emicizumab is approved for hemostatic prophylaxis for congenital hemophilia A with and without inhibitors.12,13 Anti-FVIII antibodies do not impact the efficacy of emicizumab, making it an appealing treatment for AHA. Unlike other available hemostatic options for AHA, emicizumab has a half-life of 28 days and can be administered subcutaneously.
Animal model of emicizumab in AHA and the experience in congenital hemophilia A
An early model of AHA in cynomolgus monkeys showed bleeding reduction in animals receiving weekly emicizumab (ACE910) prophylaxis.14 This study was the basis for phase 1/2 studies15 and an extensive phase 3 clinical trial program in congenital hemophilia A (HAVEN 1-4) leading to the FDA approval for prophylaxis in congenital hemophilia A.
Based on these trials, emicizumab 3 mg/kg for 4 weekly loading doses was approved for congenital hemophilia A. This results in steady state drug levels that can be maintained with 1.5 mg/kg every week, 3 mg/kg every 2 weeks, or 6 mg/kg every 4 weeks. In vitro thrombin generation studies suggest emicizumab is equivalent to an FVIII activity of over 15 IU/dL. There seems to be a ceiling effect; emicizumab does not fully correct hemostasis to that of someone without a bleeding disorder, but rather improves the clinical bleeding phenotype from severe to mild. For patients with severe congenital hemophilia A with and without inhibitors, emicizumab has revolutionized outpatient bleeding prophylaxis, resulting in significantly reduced breakthrough bleeding.12,16,17 The annual bleeding rate appears to continue to improve with longer term use of emicizumab. Additionally, emicizumab has reduced hospitalization days and the need for bypassing hemostatic therapies in patients with congenital hemophilia A and inhibitors.18
Current experience with emicizumab in people with AHA
Since the approval of emicizumab for congenital hemophilia A, there has been conceptual adaptation and use for AHA. Knoebl et al. retrospectively described 12 cases (median age 74, range 51-87 years, 50% women) of off-label use of emicizumab that showed clinical improvement of bleeding, even in patients with severe or surgical bleeding. Bleeding stopped in patients with insufficient hemostasis on bypassing agents within 4 days of emicizumab initiation, and no new or breakthrough bleeding was noted after day 2 of emicizumab therapy.19
A systematic review by Thomas et al. compiled 12 case reports with 33 patients with AHA treated with emicizumab.20 Emicizumab was initiated for active/recurring bleeding in most cases (90.9%). One patient was switched from aPCC to emicizumab after developing a myocardial infarction, and another patient was started on emicizumab to enable dual antiplatelet therapy after coronary stent placement. All reported patients had a clinical response without further spontaneous bleeding after starting emicizumab.
A retrospective survey of 87 hemophilia treatment centers in the United States in 2021 revealed that of 358 patients with AHA treated at 32 centers between 2016 and 2021, 40 patients had received off-label emicizumab. An initial look at 24 patients where data were available showed that the majority (17 of 24) were started on emicizumab to provide bleeding prophylaxis and 15 of 24 started to facilitate a transition to outpatient treatment. In 9 of 24 emicizumab was started because of failure of the current hemostatic management. After patients started emicizumab, the use of other hemostatic agents reduced from 75% to 17%, RBC transfusion from 50% to 8% and hospitalization days from a median of 10 (range 0-60) to 3 (range 0-30).21
A prospective, multicenter, open-label study of emicizumab prophylaxis in 12 adult patients led to approval of the drug for the treatment of AHA in Japan. Eleven of these patients completed the study and experienced 77 major bleeds prior to starting emicizumab. Although there were no major bleeding events, 45% (5 of 11) experienced some bleeding after emicizumab initiation. While 8 of 11 patients had used bypassing agents prior to emicizumab, only 3 patients received rFVIIa while on emicizumab, and the number of patients needing blood transfusions decreased from 7 to 3.22
Additionally, there are 2 parallel prospective multicenter trials to study the efficacy and safety of emicizumab in AHA over a 12-week period; 1 in Germany and Austria recently completed enrollment (NCT04188639), and 1 in the United States is still enrolling (NCT05345197).
Emicizumab dosing in AHA
There is no standard emicizumab dosing regimen for patients with AHA. In the retrospective case series by Knoebl et al., all 12 patients received a loading dose of 3 mg/kg weekly for 2 to 3 doses followed by maintenance dosing of 1.5 mg/kg.19 In Thomas's review of an additional 21 cases, 5 patients received the standard loading dose, where 15 had received an altered loading dose.20
A loading dose of 6 mg/kg has been entertained to reach steady state more expediently. Monthly doses of 6 mg/kg were found safe and without thrombotic complications in the HAVEN 4 study.23 A dosing regimen of 6 mg/kg on day 1 and 3 mg/kg on day 2, followed by 1.5 mg/kg weekly starting on day 8, was investigated in the prospective study in Japan and suggested steady state emicizumab concentration after 1 week.22
The impact of body mass index (BMI) on dosing of emicizumab has not been reported. While the activated partial thromboplastin time (aPTT) has been shown to correct within 1 to 3 days after initiating emicizumab,19 we have observed that normalization of the aPTT can take up to 5 days in patients with morbid obesity.
Laboratory monitoring
AHA is diagnosed and response to treatment is monitored by measuring the FVIII activity and FVIII inhibitor titer. Traditionally, this has been performed with an aPTT-based FVIII activity and the aPTT-based inhibitor assay, Nijmegen-Bethesda assay (NBA). Emicizumab interferes with the aPTT and aPTT-based assays, making traditional one-stage FVIII activity levels uninterpretable (Table 2). In a study of 12 patients with AHA, emicizumab normalized the aPTT within 1 to 3 days of administration19 even when the endogenous FVIII was still low. For patients on emicizumab, a chromogenic FVIII assay with bovine reagents must be used to monitor the patient's endogenous FVIII activity. Inhibitor titers can be measured using an ELISA-based assay, which has higher sensitivity (1.0) but lower specificity (0.83) than the NBA and is not impacted by substances that interfere with the aPTT, such as lupus anticoagulants and emicizumab.24 Currently, an emicizumab “level” is reported by some centers using the chromogenic FVIII with human reagents; however, this is not standard of care and is not recommended by the manufacturer of emicizumab for patients with congenital hemophilia A.
Considerations when choosing hemostatic agents to treat breakthrough bleeding on emicizumab
| Recombinant factor VII activated (rFVIIa) | Activated prothrombin complex concentrate (aPCC) | Recombinant porcine factor VIII (rpFVIII) | |
|---|---|---|---|
| Safety | • May increase risk of thrombosis (limited data in AHA); for congenital hemophilia A, the combination of emicizumab and rFVIIa was safe | • Thromboembolic events and thrombotic microangiopathies were reported with dose of >100 U/kg/24 hours and administered for >24 hours and the use of aPCC in people on emicizumab prophylaxis is relatively contraindicated | • Coadministration of rpFVIII and emicizumab has been reported in case series and theoretically does not have an additive effect as both compete for the same binding site |
| Limitation of use | • rFVIIa should only be reserved for acute severe breakthrough bleeding | • Avoid with emicizumab | • The chromogenic FVIII assay underestimated rpFVIII (Obizur) activity26 |
| Recombinant factor VII activated (rFVIIa) | Activated prothrombin complex concentrate (aPCC) | Recombinant porcine factor VIII (rpFVIII) | |
|---|---|---|---|
| Safety | • May increase risk of thrombosis (limited data in AHA); for congenital hemophilia A, the combination of emicizumab and rFVIIa was safe | • Thromboembolic events and thrombotic microangiopathies were reported with dose of >100 U/kg/24 hours and administered for >24 hours and the use of aPCC in people on emicizumab prophylaxis is relatively contraindicated | • Coadministration of rpFVIII and emicizumab has been reported in case series and theoretically does not have an additive effect as both compete for the same binding site |
| Limitation of use | • rFVIIa should only be reserved for acute severe breakthrough bleeding | • Avoid with emicizumab | • The chromogenic FVIII assay underestimated rpFVIII (Obizur) activity26 |
CLINICAL CASE 2
A 94-year-old man with hypertension, aortic stenosis, and a remote history of bladder cancer was diagnosed with AHA after presentation with deltoid and lower extremity hematomas. He started on off-label emicizumab with good bleeding control and improvement of the hematomas. On week 8 of treatment, he became increasingly anemic (hemoglobin 6.1 g/dL) and presented with maroon blood per rectum. He was hospitalized and transfused with 3 units of RBCs.
Treatment of breakthrough bleeding on emicizumab
While there is a clear suggestion that emicizumab may be an effective prophylactic agent to reduce subsequent or recurrent bleeding in AHA, emicizumab should not be used for acute bleeding due to its delayed bioavailability via the subcutaneous route of administration. As of now, there are no pharmacokinetic data of emicizumab associated with intravenous administration. Considering limitations for the current hemostatic agents available for AHA (Table 2), rFVIIa should probably be used over rpFVIII and aPCC for breakthrough bleeding. The combination of aPCC and emicizumab carries a black box warning due to the risk of thrombotic events and thrombotic microangiopathy seen in trials of congenital hemophilia A. Combining emicizumab with rFVIIa 90 mg/kg appears safe and in vitro data25 suggest higher doses may be feasible, but thromboembolic complications have been reported.
Laboratory monitoring of rpFVIII limits its use with emicizumab. rpFVIII is an effective hemostatic option in AHA6 and can be monitored with the one-stage, aPTT-based FVIII clotting assay, which is uninterpretable in patients on emicizumab. The chromogenic FVIII assay was shown to underestimate rpFVIII (Obizur) activity.26 Considering that the one-stage assay is unreliable in patients on emicizumab, monitoring of FVIII activity for rpFVIII therapy is challenging. Concurrent use of rpFVIII with emicizumab can be considered for acute bleeding, with the caveat that a target chromogenic FVIII activity has not been established, since it gives falsely low results (underestimates) with rpFVIII.
CLINICAL CASE 2 (continued)
The patient continued to have GI bleeding, and a decision was made to add tranexamic acid to his hemostatic management. However, he continued to have GI bleeding requiring transfusions, for which rFVIIa 90 µg/kg was initiated. He received 14 doses of rFVIIa over 6 days and then developed acute right-sided ataxia. rFVIIa was discontinued and his stoke symptoms resolved without residual deficits. A capsule endoscopy demonstrated telangiectasias in mid jejunum/proximal ileum for which he started octreotide with good bleeding control.
Adverse events of special interest
Common adverse events to emicizumab include injection site reactions, headaches, and arthralgias. Additional adverse events must be considered in patients with AHA on emicizumab given the high rate of comorbidities and advanced age of this population. AHA is a condition associated with high morbidity and mortality, likely due to the advanced age of patients, their underlying comorbidities, the use of IST, and, less often, bleeding.10,27 As would be expected in this population, thromboembolic events, especially when bypassing agents are used, are not uncommon despite the AHA state.10
Thromboembolism
In the case series by Knoebl et al., a 79-year-old experienced a minor stroke on day 16 of emicizumab after receiving rFVIIa 90 µg/kg prior to a procedure.19 Of the 24 patients at the US centers, 1 developed a lower extremity deep vein thrombosis (DVT) while on weekly emicizumab therapy.21 The prospective Japanese study of 12 patients reported 1 event of an incidental lower extremity DVT on day 16 of emicizumab treatment that resolved without treatment.22
From our experience so far, we would anticipate that breakthrough bleeding can occur on emicizumab and that patients need additional hemostatic prophylaxis for invasive procedures. From the early experience during the HAVEN 1 study, we know that the use of aPCC is contraindicated in patients on emicizumab and can lead to thrombosis and thrombotic microangiopathies.12 The coadministration of emicizumab and rFVIIa, however, was safe in people with congenital hemophilia A.28 Prospective assessment of thromboembolic events for people receiving emicizumab for AHA is ongoing in the above-mentioned parallel clinical studies.
Anti-drug antibodies
Data from the congenital hemophilia studies showed that 5.1% (34 of 668) of people exposed to emicizumab developed anti-emicizumab antibodies. Of these antidrug antibodies, 41.2% were transient, 52.9% were neutralizing in vitro, and only 0.6% resulted in decreased emicizumab plasma levels.29 In the prospective Japanese study, 1 of 12 (8.3%) developed an antidrug antibody, but it did not have a clear impact on the pharmacokinetics.22
CLINICAL CASE 3
A 76-year-old man with poorly controlled hypertension and diabetes had prolonged aPTT for at least 3 years. He had no abnormal bleeding and was never further evaluated or treated for his prolonged aPTT. He then emergently presented with an incarcerated inguinal hernia that required surgery. He was diagnosed with AHA with an FVIII activity of 5 IU/dL and FVIII inhibitor of 11.9 BU. He received aPCC perioperatively and did well. Postoperatively, he was seen in clinic and started on cyclophosphamide 100 mg PO daily for immunosuppression. He was felt to be a poor candidate for high-dose corticosteroid due to his history of hypertension and diabetes. Fourteen days later, he presented to the emergency department with nausea and ankle swelling and was found to be hyponatremic with sodium levels of 115 mmol/L (normal 136-145 mmol/L) with mildly elevated liver enzymes. Cyclophosphamide was held and he received supportive care with resolution of the hyponatremia and transaminitis, but 5 days later he developed massive, spontaneous bilateral iliopsoas bleeding resulting in bilateral leg weakness and loss of sensation in both anterior thighs, profoundly impacting ambulation and self-care.
Impact of emicizumab on immunosuppression
While approximately 10% of patients with AHA do not initially present with bleeding,7 a lack of bleeding history does not negate future bleeding risk or eliminate the need for immunosuppression. Historically, early use of immunosuppression for AHA was essential to eradicate the inhibitor and control bleeding. However, immunosuppression-related adverse events such as infections are a main driver of the high morbidity and mortality of AHA. With effective outpatient bleeding prophylaxis, emicizumab reduces the urgency to eradicate the inhibitor, allowing reduced-intensity immunosuppression. In a survey of adult hematologists at 87 US hemophilia treatment centers, 70% of providers with experience with emicizumab for AHA reported using emicizumab to delay or decrease immunosuppression.21 In Knoebl's case series, all patients started on emicizumab were deemed to be poor candidates for immunosuppression.19 The safety of postponing immunosuppression with emicizumab may be answered in a planned analysis between the 2 ongoing clinical trials in Europe (NCT04188639) with delayed initiation of IST and the United States (NCT05345197) with standard initiation of IST.
CLINICAL CASE 3 (continued)
The patient required several RBC transfusions and was treated with rFVIIa initially and then aPCC with stabilization. He was not able to care for himself at home and needed in-patient rehabilitation, which proved difficult because he continued on aPCC every 12 hours to prevent further bleeding. Further, the next line IST, rituximab, was difficult to administer in the rehab setting. Considering the ongoing need for a bleeding prophylaxis that could be given in rehab as well as a potential delay in further IST, he was started on off-label emicizumab with an accelerated loading dose. This allowed for transition to rehab, where he did not have recurrent bleeding and regained strength, allowing for transition to home 2 months later. He then received rituximab 100 mg weekly × 4 doses and cleared his inhibitor 6 weeks later.
Duration of emicizumab use in AHA
Monitoring patients with AHA for partial and complete remission of the inhibitor is essential and will guide the duration of emicizumab therapy. Laboratory monitoring on emicizumab was discussed by Platton et al. in an earlier chapter and has special considerations (Table 3). Emicizumab may be discontinued once the endogenous FVIII level improves; however, there is no strict FVIII threshold for discontinuing emicizumab and there is a variety in clinical practice: emicizumab was discontinued when FVIII ranged from 10% to 86% in 26 patients with AHA in a systematic review by Thomas et al.20 In our practice, we continue emicizumab if there is still a persistent inhibitor and low FVIII level, especially if the patient has a history of bleeding as it can recur without prophylaxis. Data from 2 prospective clinical trials in Europe (NCT04188639) and the United States (NCT05345197) may help guide emicizumab dosing and stopping thresholds. Due to the long half-life of emicizumab, some effect of the drug can likely be anticipated until 5 months after discontinuation.
Impact of emicizumab on labs
| Lab | Impact of emicizumab | Recommended alternative |
|---|---|---|
| aPTT | False decrease | Do not use while on emicizumab |
| One-stage FVIII (PTT based)* | False increase | Chromogenic FVIII with bovine reagents (measure infused and/or endogenous FVIII) |
| FVIII inhibitor titer with clotting-based assays (Bethesda) | Uninterpretable | Chromogenic Bethesda assay with bovine reagents |
| Activated clotting time (ACT) | False decrease | Anti-Xa activity |
| Activated protein C resistance (aPTT based) | Uninterpretable | Factor V Leiden genetic testing (if clinically appropriate) |
| Lab | Impact of emicizumab | Recommended alternative |
|---|---|---|
| aPTT | False decrease | Do not use while on emicizumab |
| One-stage FVIII (PTT based)* | False increase | Chromogenic FVIII with bovine reagents (measure infused and/or endogenous FVIII) |
| FVIII inhibitor titer with clotting-based assays (Bethesda) | Uninterpretable | Chromogenic Bethesda assay with bovine reagents |
| Activated clotting time (ACT) | False decrease | Anti-Xa activity |
| Activated protein C resistance (aPTT based) | Uninterpretable | Factor V Leiden genetic testing (if clinically appropriate) |
The one-stage FVIII activity will overestimate the impact of emicizumab and will not be able to separate the endogenous FVIII activity from the impact of emicizumab on the assay. Emicizumab impacts all single-factor assays that are performed with one-stage, aPTT-based assays (such as factor IX activity).
Conclusion
Although currently off label for AHA, emicizumab is poised to revolutionize treatment through effective bleeding prophylaxis that can be given in the outpatient setting, reducing hospitalizations and the need for bypassing agents. Unlike rpFVIII, the FVIII autoantibodies do not impact the use of emicizumab, and antidrug antibodies are rare. By reducing the risk of bleeding, emicizumab may shift the treatment goals of AHA away from high-dose immunosuppression, reducing the risk of adverse events such as infections. Emicizumab cannot be used for breakthrough bleeding, and care must still be taken to monitor for thrombotic events with concurrent use of bypassing agents, especially given the high rate of comorbidities in the AHA population. Two ongoing prospective parallel studies in Europe and the United States will answer important questions about the efficacy and safety of emicizumab in AHA.
Conflict-of-interest disclosure
Jacqueline Poston is a consultant for TeraImmune.
Rebecca Kruse-Jarres is an educational speaker for Genentech, is a scientific advisor for Regeneron and Roche, and received research funding from Genentech.
Off-label drug use
Jacqueline Poston: Off-label use of emicizumab was discussed.
Rebecca Kruse-Jarres: Off-label use of emicizumab was discussed.
