

Abstract
Curative therapies for sickle cell disease include allogeneic hematopoietic stem cell transplantation (HSCT) and gene-modified autologous stem cell transplantation. HSCT has been used for 30 years with success measured by engraftment, symptom control, graft-vs-host disease (GVHD) risk, organ toxicity, and immune reconstitution. While human leukocyte antigen–matched sibling donor (MSD) transplants have excellent outcomes, alternate donor transplants (unrelated/haploidentical) are just beginning to overcome GVHD and engraftment hurdles to match MSD. Gene therapy, a newly developed treatment, is undergoing careful evaluation in many trials with varying approaches. The risk/benefit ratio to the patient in relation to outcomes, toxicities, and mortality risk drives eligibility for curative interventions. Consequently, eligibility criteria for MSD transplants can be less stringent, especially in the young. Posttransplant outcome analysis after the “cure” with respect to organ function recovery is essential. While established damage such as stroke is irreversible, transplant can help stabilize (pulmonary function), prevent further deterioration (stroke), improve (neurocognition), and protect unaffected organs. Tracking organ functions postintervention uniformly between clinical trials and for adequate duration is essential to answer safety and efficacy questions related to curative therapies. Age-appropriate application/outcome analyses of such therapies will be the ultimate goal in overcoming this disease.
Learning Objectives
Apply knowledge regarding the current status of curative therapy interventions to determine indications and eligibility
Track organ functions across clinical trials following curative therapies to determine components and extent of cure
CLINICAL CASES 1A AND 1B
“George” was 12 years old with hemoglobin Sβ+ thalassemia. He developed silent cerebral infarcts and acute chest syndrome (ACS) × 2, and he had pain-related admissions impairing school attendance/performance despite hydroxyurea therapy. His hemoglobin was 7.5 g/dL. Hemoglobin F was 8.6%.
His 6-year-old brother with similar hemoglobinopathy had occasional fevers and 1 mild ACS. He was on hydroxyurea therapy. His hemoglobin was 7.2 g/dL; hemoglobin F was 7.8%.
After extensive discussion regarding treatment and natural history, per family preference, they underwent reduced intensity conditioning (RIC) and hematopoietic stem cell transplantation (HSCT) 1 year apart following peripheral blood stem cell collection once from their 10-year-old human leukocyte antigen–matched sibling donor (MSD) with sickle trait.
Now 5 and 4 years post-HSCT, respectively, neither has disease-related symptoms or hemolysis, and both have hemoglobin levels of 13.5 g/dL. George's brain magnetic resonance imaging continues to have the preexisting frontal T2/fluid attenuated inversion recovery (FLAIR) hyperintensity. He has an Individualized Education Plan in school but is grade appropriate.
CLINICAL CASE 2
“Adam,” with hemoglobin SS disease, was 21 years old when he decided to consider HSCT. He has been on chronic red cell transfusion therapy (CRTT) since having an ischemic stroke at 17 months of age and chelation therapy since 4 years of age. He had a right thalamic infarct, left-sided hemiparesis that improved gradually, and learning difficulties in school. He underwent a matched unrelated donor RIC transplant and weaned systemic graft-vs-host disease (GVHD) prophylaxis at 1.5 years. The thalamic infarct remains unchanged 6.5 years post-HSCT. He completed law school and is employed. Pre-HSCT, he had severe c-holelithiasis/pancreatitis. Post-HCT, he has mild pancreatic insufficiency and continues enzyme replacement. Hemoglobin/hemolytic parameters are normal, as are other organ functions.
These cases represent the complex age and symptom variability of the sickle cell disease (SCD) phenotype. The pathology of target organ damage is similar although at a variable pace and severity. Table 1 shows the extensive nature of vasculopathy and organs involved. The most frequent, muscular vaso-occlusive pain episodes (VOEs) are reversible with adequate medical management. Symptom frequency and intensity impair physical and mental well-being and quality of life. Vasculopathy involving vital organs can cause irreversible sequelae, morbidity, and premature mortality.1 Recently approved medications decrease red cell polymerization and endothelial damage to decrease the frequency/severity of VOEs, but long-term benefits on organs are unknown. Curative interventions (allogeneic HSCT/autologous gene-modified HSCT) can change disease course. Safety and efficacy signals are better known for the former and under investigation in the latter.
The spectrum of sickle cell disease manifestations and effects of transplant
| Organ | Pathology | Clinical manifestations | Effect of transplant |
|---|---|---|---|
| Blood and vessels | Red cell polymerization Red cell shape distortion ↓ membrane fluidity Abnormal rheology Red cell–leukocyte–endothelial adhesive interaction Oxidative stress | Hemolysis Chronic anemia Inflammation Infarction | Reversed |
| Brain | Stenosis Velocity perfusion imbalance Vessel beading Moyamoya | Posterior reversible encephalopathy syndrome Ischemic stroke Hemorrhagic stroke Silent infarcts | Improved or stabilized |
| Cardiopulmonary | Hemolysis Chronic anemia Ischemia reperfusion injury Red cell polymerization | Increased left ventricular mass Left ventricular diastolic dysfunction Pulmonary hypertension Acute chest syndrome Obstructive lung disease Restrictive lung disease | Improved or stabilized |
| Liver | Red cell polymerization Hemolysis Hyperbilirubinemia Iron accumulation | Sickle hepatopathy Transfusional hemosiderosis Biliary disease Hepatic fibrosis/cirrhosis | Improved or reversed |
| Spleen | Congestion, hemolysis Infarction | Splenic crisis and congestion Autosplenectomy | Improved or stabilized |
| Kidneys | Intramedullary sickling Papillary necrosis Hyperfiltration | Hematuria Acute kidney injury Chronic kidney disease Renal failure | Improved or reversed |
| Gonads | Iron deposition Microvascular occlusion Ovarian and testicular infarction | Urinary tract infection Dysmenorrhea Low ovarian reserve Priapism Low sperm count | Stabilized |
| Bone and joints | Perfusion impediment | Osteonecrosis Avascular necrosis | Progression halted |
| Muscles | Perfusion impediment | Vaso-occlusive episodes | Reversed |
| Organ | Pathology | Clinical manifestations | Effect of transplant |
|---|---|---|---|
| Blood and vessels | Red cell polymerization Red cell shape distortion ↓ membrane fluidity Abnormal rheology Red cell–leukocyte–endothelial adhesive interaction Oxidative stress | Hemolysis Chronic anemia Inflammation Infarction | Reversed |
| Brain | Stenosis Velocity perfusion imbalance Vessel beading Moyamoya | Posterior reversible encephalopathy syndrome Ischemic stroke Hemorrhagic stroke Silent infarcts | Improved or stabilized |
| Cardiopulmonary | Hemolysis Chronic anemia Ischemia reperfusion injury Red cell polymerization | Increased left ventricular mass Left ventricular diastolic dysfunction Pulmonary hypertension Acute chest syndrome Obstructive lung disease Restrictive lung disease | Improved or stabilized |
| Liver | Red cell polymerization Hemolysis Hyperbilirubinemia Iron accumulation | Sickle hepatopathy Transfusional hemosiderosis Biliary disease Hepatic fibrosis/cirrhosis | Improved or reversed |
| Spleen | Congestion, hemolysis Infarction | Splenic crisis and congestion Autosplenectomy | Improved or stabilized |
| Kidneys | Intramedullary sickling Papillary necrosis Hyperfiltration | Hematuria Acute kidney injury Chronic kidney disease Renal failure | Improved or reversed |
| Gonads | Iron deposition Microvascular occlusion Ovarian and testicular infarction | Urinary tract infection Dysmenorrhea Low ovarian reserve Priapism Low sperm count | Stabilized |
| Bone and joints | Perfusion impediment | Osteonecrosis Avascular necrosis | Progression halted |
| Muscles | Perfusion impediment | Vaso-occlusive episodes | Reversed |
Indications for curative therapy should account for the risk/benefit ratio. Modern supportive care efficiently prevents SCD-related mortality during childhood in high-income countries. Childhood survival is 93% to 98% depending on disease severity.2 Similarly, registry databases and clinical trial outcome data are available for allogeneic HSCT. These allow outcome tracking/toxicities/reasons for failure and comparison between donor sources and transplant methods. With improving results for cure (>90% disease-free survival in recent reports),3-5 indications for curative interventions will continue to evolve.6 An example is “less severe” disease and HSCT in young patients (<13 years) with MSD.7 It also provides impetus for trials expanding donor options (<25% have an MSD), improving conditioning regimens, and incorporating novel GVHD prophylaxis. Gene therapy averts GVHD risk/immune suppression. Higher-risk transplants (older patients, alternative/mismatched donors, preexisting organ dysfunction) require careful eligibility consideration and care. Acceptable indications for all transplants include damaging disease sequelae such as central nervous system (CNS) ischemia/vasculopathy, recurrent ACS, severe/recurrent VOEs, or mortality predictors such as pulmonary hypertension. Morbidity indicators include CRTT and red cell alloimmunization. Inclusions of nephropathy/retinopathy stem from newer natural history studies. Prevention/progression is independent of reversibility. Figure 1 shows the definitive and “gray” zones of eligibility based on disease risks and HSCT benefits, as highlighted in SCD manifestation differences between George, his brother, and Adam. Current trials of gene therapy include patients 5 to 45 years of age with severe symptoms that justify experimental intervention in the absence of MSD. However, the requirement for chemoablation prior to gene therapy can limit this option for those with severe organ function impairment.
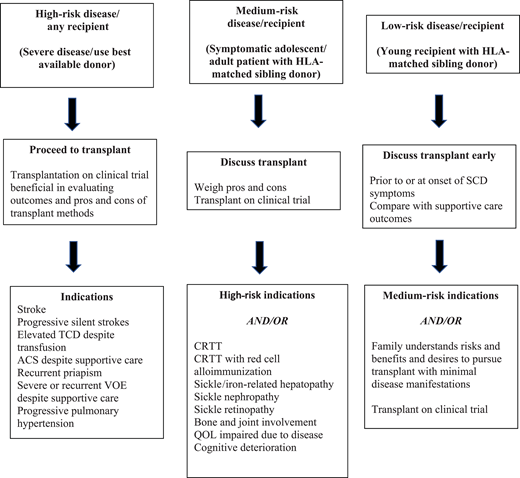
Expanding transplant indications based on risk/benefit ratio. HLA, human leukocyte antigen; QOL, quality of life; TCD, transcranial Doppler velocity.
Expanding transplant indications based on risk/benefit ratio. HLA, human leukocyte antigen; QOL, quality of life; TCD, transcranial Doppler velocity.
Early advantages following curative therapies
HSCT reverses red cell polymerization and eliminates red cell–leukocyte–endothelial cell damage, oxidative stress, and inflammation. Those with unremitting pain despite conservative management are good candidates for curative therapy.8 Successful HSCT eliminates pain early in children and gradually in adults.9 Reversibility is best noted in patients with acute episodic pain that remits with short-acting opioids, as in the case of George. Chronic pain, as well as that requiring prolonged long-acting opioid therapy, is more difficult. In a cohort of 20 adult patients who underwent a nonmyeloablative matched sibling transplant, risk factors such as older age, more severe and persistent pain before HSCT, and more symptoms of anxiety were associated with persistent pain following transplant.10 Pain control, when achieved, decreases hospital admissions and opioid use. Functional and performance scores, as well as health- related quality of life, improve in the months following and are evident 1 year posttransplant in children following MSD HSCT.11 This improvement in health, health perception, self-esteem, and function was evident in unrelated donor HSCT despite GVHD, following haploidentical transplants, and in adults with established pain prior to HSCT, although there is interindividual variation, more so in adults.12-15 Fatigue remained a lingering issue in adults following myeloablative HSCT.13 Elimination of pain is evident after gene therapy and is a primary outcome measure.16
Individuals with preexisting end-organ damage/vasculopathy are susceptible to transplant complications—hypertension, consequent posterior reversible encephalopathy syndrome (PRES), and hemorrhagic infarcts due to CNS vasculopathy (compounded by calcineurin inhibitors/steroid therapy/thrombocytopenia). Hence, nuanced supportive care, including transfusions to suppress hemoglobin S pre-HSCT, hemoglobin levels maintained in the low-normal range (9-11 g/dL) to avoid hyperviscosity, aggressive hypertension control (<90th percentile for age), and thrombocytopenia prevention (platelets >50 × 109/L), are important. While PRES suggests reversibility, persistent white matter changes and neurologic dysfunction may persist. Modified preparative regimens and supportive care strategies have decreased PRES. A PRES incidence of 11% to 34% decreased to 7% after eliminating corticosteroids, shortening exposure to calcineurin inhibitors, or substituting with sirolimus.4,12 Modern GHVD prophylaxis strategies that include abatacept or cyclophosphamide result in excellent PRES control.
Brain involvement
Both George and Adam presented with SCD-related involvement of the brain in varying severity. Small-caliber brain vessels are common vasculopathy targets that manifest as stenosis, beading, moyamoya, increased flow velocity, perfusion imbalance, and eventually overt ischemic or hemorrhagic stroke or silent infarctions. Historically, overt stroke was an established indication for HSCT, given the CRTT for secondary stroke prevention and the incidence of breakthrough despite this.17 Although control of progression of vascular damage is the goal, the brain is also a susceptible site for ischemia or intracerebral hemorrhage during HSCT or in the immediate post-HSCT period due to the inflammatory/thrombocytopenic milieu. Cerebral vasculopathy, acute complications that alter blood oxygen content, and cerebral blood flow autoregulatory exhaustion are thought to contribute to complications. A retrospective study performed in an SCD transplant consortium found that the risk of neurologic complications post-HSCT was higher with pre-HSCT brain involvement.18
HSCT provides better neuroprotection than CRTT. Following myeloablative MSD HSCT, stroke recurrence was noted in 2 of 36 recipients with pre-HSCT stroke.19 Subsequent reports in >60 patients demonstrated no strokes after ablative or RIC HSCT and good supportive care.20,21 In contrast, 10% to 20% of patients developed strokes despite continuing CRTT.17,22,23
Strokes and silent cerebral infarcts represent fixed injury. While HSCT prevents damage extension, established lesions are seldom reversible, making a case for predamage intervention. MSD HSCT significantly decreases transcranial Doppler velocities compared with standard of care by 1 year posttransplant.24 Improvement in dynamic measures of brain injury such as axonal integrity and cerebral metabolic stress normalization can also be demonstrated post-HSCT.25-27 Both George and Adam had radiologic CNS stability posttransplant. Furthermore, in a series of 10 patients who underwent transplantation from MSD or alternate donors, abnormal cerebral blood flows and oxygen extraction fractions returned to normal non-SCD levels 12 to 24 months post-HSCT, a result better than that noted in children receiving chronic red cell transfusion therapy.28
Neurocognition
Cognitive deficits are common among children and adults, with some detected before 1 year of age.29,30 Intelligence quotient (IQ) is projected to decline with age, and domain-specific deficits in executive function, attention, and processing speed are prevalent.31-33 Given the association of cognition with anemia and oxygen saturation, a permanent increase in hemoglobin with less sickling should result in stable to improved cognitive function.
Brain imaging is standard in medical centers performing curative interventions. Neuropsychological assessment is more challenging. In the SCURT trial, a secondary outcome included a battery of neuropsychological tests—IQ and several domain-specific measures. Only 13 of 29 children completed IQ assessments before and 2 years post-HSCT. The IQ post-RIC and unrelated donor HSCT was stable after 2 years.34 In a single- institution experience of 9 children who underwent MSD HSCT, IQ before and 3 years after MSD was stable.35 Another cohort of 16 children and adolescents who underwent haploidentical HSCT also demonstrated stable IQ and improved processing speed after 1 year.36 A French cohort (15 children) had the longest follow-up of 5 years post-MSD HSCT. Stable IQ and improved processing speed were demonstrated.37 Taken together, HSCT recipients consistently demonstrate stabilized/improved cognition. Again, HSCT could be more protective at younger ages prior to the development of vasculopathy. To determine the scope of treatment, it is imperative that domain-specific measures of cognition are included in trials as an aspect of the risks and benefits of intervention.38
Kidney
Acute and chronic kidney disease evolves with age. Hyperfiltration and microalbuminuria begin in childhood. Acute complications are frequently accompanied by acute kidney injury. Nonsteroidal anti-inflammatory analgesics used for VOE pain can contribute to chronic kidney disease. By the time HSCT is considered, many patients have clinical or subclinical sickle nephropathy. In 18 children with SCD, hyperfiltration was present in 44% pre-HSCT but declined to 22% at 2 years post-HSCT.39 However, one-third developed hypertension, likely exacerbated by nephrotoxic transplant medications, compared with an incidence of 5.6% pre-HSCT. Adults with SCD have a 12% prevalence of renal failure by 37 years of age, an irreversible change contributing to morbidity and mortality.40 However, peri-HSCT, acute kidney injury was noted early in 58% of recipients, 10% of which was severe (stage 3), again likely due to nephrotoxic agents used early post-HSCT. Thus, minimizing nephrotoxic agents coupled with transplant-mediated control of vaso-occlusion within the acidic/osmolar renal medulla by HSCT can aid preservation of renal function.
Liver
Sickle-related hepatic disease can include asymptomatic biliary dysfunction, acute hepatic crisis ± red blood cell sequestration, hyperbilirubinemia, hepatocellular injury, and synthetic function failure, a consequence of vaso-occlusion in hepatic sinusoids and liver parenchyma/terminal bile duct ischemia.41,42 Hepatic iron overload and cirrhosis contribute to chronic liver disease. In a single-center review, 10% of adult SCD deaths were attributed to cirrhosis.43 Post-HSCT, gallstones and consequent pancreatitis should decrease due to the elimination of hemolysis and hyperbilirubinemia. In our case 2, pre-HSCT choledochopancreatic compromise was preexisting, and pancreatic insufficiency manifested after, suggesting compromised reserve. The general theme of preexisting pathology (SCD and iron related), HSCT-induced toxicity such as sinusoidal obstructive syndrome (SOS) necessitates awareness, monitoring, and intervention. Although less of a concern with SCD, hepatic iron with bridging fibrosis is a risk for SOS and graft failure in thalassemia. Adolescents and adults treated with a reduced-intensity (fludarabine/melphalan) or reduced-toxicity (busulfan/fludarabine) regimen did not develop SOS.13,21 HSCT should protect against vaso-occlusion–induced hepatic crisis. Serum ferritin levels decrease following successful transplant and cessation of transfusions, although a preexisting iron load may need to be tackled with phlebotomy or post-HSCT chelation.44 The successful recovery to normal hemoglobin levels and eradication of hemolysis following gene therapy should also offset iron-related pathology in similar manner.45
Cardiopulmonary benefits
Life span is compromised by SCD, with a spike in mortality at 35 to 44 years.1 Cardiopulmonary pathology is the commonest culprit.46 Pathology commences during late childhood/adolescence and includes ACS/pneumonia, pulmonary hypertension, systemic hypertension, congestive cardiac failure, left ventricular diastolic dysfunction, myocardial infarction, and arrhythmia. A higher tricuspid regurgitation jet velocity (>3.1 m/s) and lower hemoglobin (< 8.3 g/dL) were mortality predictors.47,48 HSCT can avoid cardiopulmonary deterioration if accomplished before established pathology. ACS risks receded posttransplant; stability or improvement was noted in restrictive and obstructive pulmonary disease in most patients, measured at 2 years post-HSCT with pulmonary function tests (forced expiratory volume, forced expiratory volume in the first second/forced vital capacity, total lung capacity, and diffusing capacity of the lungs for carbon monoxide).49,50 Similarly, cardiac size and diastolic filling continued to improve in the first year, with a trend toward improvement in the 6-minute walk time.51 Twenty- four patients from this group had a high tricuspid regurgitation velocity (a measure of increased pulmonary pressure and a mortality predictor) at a median of 2.7 m/s, which decreased to 2.3 m/s at 1 year post-HSCT, indicating benefit. These functions, however, continue to require diligent follow-up after curative therapy long term.
Summary
Curative therapies for SCD commenced with ablative transplant strategies similar to hematopoietic malignancies about 4 decades ago and have evolved since. Clinical trials have targeted lower-intensity/toxicity conditioning, expanded donor sources to unrelated marrow, expanded cord and haploidentical products, modified GVHD prophylaxis, and enhanced supportive care and monitoring to gradually but definitively improve outcomes and increase HSCT access. Consequently, eligibility for transplant has expanded for indications and age. This expansion results in variable pre-HSCT disease baseline and thus variable outcomes post-HSCT ranging from complete elimination of disease manifestations in the young to stemming progression of complications in older recipients. Baseline disease status also determines time to recovery post-HSCT. The trajectory of recovery is likely to differ with gene therapy approaches as well. As transplant and gene therapy trials proliferate, defining outcomes and success demand uniformity in assessment and compartmentalization based on recipient status. Outcome guidelines have been developed within the American Society of Hematology and transplant consortia such as the Pediatric Transplant and Cellular Therapy Consortium.38,52,53 It is also imperative to track recipients long term to determine late outcomes, and formal efforts to achieve this are now under way.
Conflict-of-interest disclosure
Monica L. Hulbert is a consultant for Bluebird Bio and Global Blood Therapeutics, has received research funding from Forma Therapeutics and Global Blood Therapeutics, and has family member employment with Pfizer Inc.
Allison A. King has received research funding from Global Blood Therapeutics.
Shalini Shenoy is vice chair on the ASH publications committee, DSMB chair with Aruvant Technologies, a member of NHLBI DSMB, and attended the advisory board meeting for Bristol Myers Squibb.
Off-label drug use
Monica L. Hulbert: nothing to disclose.
Allison A. King: nothing to disclose.
Shalini Shenoy: nothing to disclose.
