

Abstract
Enthusiasm about interferons for the treatment of myeloproliferative neoplasms has recently arisen. How does a nontargeted therapy selectively target the malignant clone? Many foundational questions about interferon treatment are unanswered, including who, when, and for how long do we treat. Using an individual case, this review touches on gaps in risk assessment in polycythemia vera (PV) and essential thrombocythemia (ET) and the history of treatment with interferons. How is it that this proinflammatory cytokine effectively treats ET and PV, themselves proinflammatory states? We summarize existing mechanistic and clinical data, the molecular context as a modifier for treatment response, the establishment of treatment goals, and the challenges that lie ahead.
Learning Objectives
Define treatment goals in PV/ET
Summarize molecular responses using IFN in PV/ET
CLINICAL CASE
A 32-year-old woman with at least 2 years of blood count abnormalities is referred to hematology. Two years prior, early in a complicated pregnancy, she had an abnormal complete blood count: white blood cell count, 12.7 thou/µL; hemoglobin, 16 g/dL; mean corpuscular volume, 82 fL; and platelets, 415 thou/µL. She was admitted with preeclampsia 25 weeks into her pregnancy and was closely observed, treated with low-dose aspirin, and delivered at 32 weeks. Her newborn daughter was born small for gestational age. When first evaluated by hematology 19 months later, she described chronic itching for 7 to 8 years, worse after a shower and absent during pregnancy but present again and mild and manageable. She had no visual symptoms or headaches and no history of clots. Medications included an oral contraceptive. A repeat complete blood count showed persistent abnormalities: WBC, 11.3 tho/µL; hemoglobin, 17.9 g/dL; mean corpuscular volume, 76 fL; platelets, 692 tho/µL. Additional testing, including JAK2V617F mutation analysis, detected a mutation at an allele frequency of 32.6%. Serum ferritin was 7 ng/mL, and serum erythropoietin was 2 mIU/mL (range, 3-19 mIU/mL), confirming a diagnosis of polycythemia vera (PV). While her symptoms now feel mild and manageable, she is concerned about the overall prognosis of her disease and wonders about the safety of future pregnancies, which she desires.
Introduction
Our patient has PV, and her experience is common to many when diagnosed: long-standing but nonspecific symptoms and a lag from the time of blood count abnormalities to confirmation of diagnosis. More pernicious is her experience in pregnancy, fraught with complications that are too common in women with PV, including the overlooked real-time diagnosis. Nevertheless, by standard vascular risk stratification criteria, our patient is considered to have low-risk disease, a designation of little comfort to many young people with myeloproliferative neoplasms (MPN). How can we best address her immediate concerns related to thriving as a “thirty-something,” the intermediate goal of growing her family, and, finally, the long-term and often dominant concern related to progression of disease and early mortality? Many are hopeful that interferons (IFNs), especially long-acting formulations, might address such patient concerns.
MPN as inflammatory diseases
Classical MPN without the Philadelphia-negative chromosome are clonal diseases classified into 3 subtypes: PV, essential thrombocythemia (ET), and primary myelofibrosis (PMF). They are clinically characterized by proliferative bone marrow, abnormal blood counts, splenomegaly, and constitutional symptoms, along with a long-term higher risk of secondary bone marrow fibrosis and acute leukemic transformation. In 2005 the 9 oncogenic driver mutation was identified and found to be present in nearly all patients with PV and about 50% to 60% of patients with ET or PMF. Activating mutations in thrombopoietin receptor (MPL) and calreticulin (CALR) account for the large majority of remaining cases. These driver mutations functionally converge in a common pathway, resulting in aberrant JAK2-STAT signaling.
The genetic drivers of MPN converge, with growing evidence that proinflammatory processes are central to MPN pathobiology. MPN are acquired diseases of stem cells, and the JAK2 pathway is considered an important inflammatory driver. Patients with MPN have increased levels of inflammatory cytokines, dysregulation of immune-related genes, and enhanced oxidative stress.1,2 As of the summer of 2021, JAK inhibitors remain the only approved medications in the United States for PV and PMF, and central to their activity is the ability to modulate cytokine profiles and improve symptoms. Despite off-label use, IFNs predated JAK inhibitors for the treatment of MPN. IFNs have been considered a standard treatment for decades, predating even the identification of the driver mutations. But we are now left with a central paradox of treating a proinflammatory state with a proinflammatory cytokine. Optimally, the experience of IFN use and mechanistic preclinical and clinical data will lead us on a path to resolution of this paradox and, more importantly, improved outcomes for our patients.
History of IFN use as it relates to the treatment of PV and ET
Let us now consider treating our patient. Recombinant IFN-alpha has been a treatment option for patients with hematologic malignancies for decades and was first used to treat MPN over 30 years ago. In the pre–tyrosine kinase era, IFN was able to induce deep remissions in a small subset of patients with chronic myeloid leukemia, while conventional chemotherapies failed.3 In Philadelphia chromosome-negative MPN, an early study reported on 12 patients with improvement in thrombocytosis with 2 to 10 weeks of recombinant IFN.4,5 Despite other early trials showing promising responses, the side effects with IFN forced unacceptable discontinuation rates, and consequently, its use was not widely adopted. MPN patients can experience debilitating constitutional symptoms, felt to be cytokine driven, making expected but overlapping IFN-related side effects potentially even less well tolerated in this population. The development of pegylated IFNs (pegIFNs), a longer-acting formulation that is better tolerated and requires less frequent dosing, renewed interest in this treatment approach. More recent studies, summarized here, have shown promising responses in hematologic abnormalities, symptoms, and anticlonal activity with the use of pegIFN.
The exact action of IFN in MPN remains an area of intense investigation. Supported mechanisms include direct cytotoxic effects on the malignant clonal cells in addition to enhancing the host innate and adaptive immune response. IFN may increase the expression of neoantigens on malignant clonal cells, rendering them more susceptible to immunosurveillance,6 and it can also induce emergence of the hematopoietic stem cell from quiescence to allow for apoptosis (extensively reviewed in Kiladjian, Mesa, and Hoffman et al).7 Ideally, correlative science from prospective studies of peg-IFN can help elucidate the mechanisms most relevant for optimal response.
Goals of therapy and risk stratification
The goals of treating patients with MPN include amelioration of symptoms, reduction in thrombosis risk, control of abnormal blood counts, reduction in long-term consequences, and, hopefully, delay of disease progression. In addition to an often dominant concern for progression to advanced myelofibrosis and/or acute leukemia, additional consequences of MPN therapies can accumulate over time; thus, thoughtful treatment recommendations are crucial.
Therapeutic phlebotomy (TP) alone was found to be superior to alkylating agents in one of the early pivotal studies of interventions for PV, is the backbone of treatment in newly diagnosed PV, and persists as an important Hippocratic reminder that simple can be better.8 TP is particularly critical in the acute setting at diagnosis when signs or symptoms of hyperviscosity are present. A more recent foundational study, CYTO-PV, confirmed that hematocrit control below 45%, by means of TP or cytoreductive medications, reduced the risk of vascular events by approximately 4-fold.9 But TP only controls red blood cell count, not platelets and white blood cells, so proliferative disease is only partially addressed with this satisfyingly simple and safe intervention. In addition, iron deficiency—sometimes present even prior to phlebotomy initiation in PV patients such as ours—is induced or worsened by TP, which often drives platelet counts higher still and in aggregate may cause or exacerbate microvascular or neurocognitive symptoms. PegIFN, by contrast, induces trilineage blood cell count lowering, and when responses are seen, it can offer the possibility of maintaining blood count control while still correcting iron deficiency, thus interrupting the vicious TP-iron deficiency-thrombocytosis cycle (Figure 1).
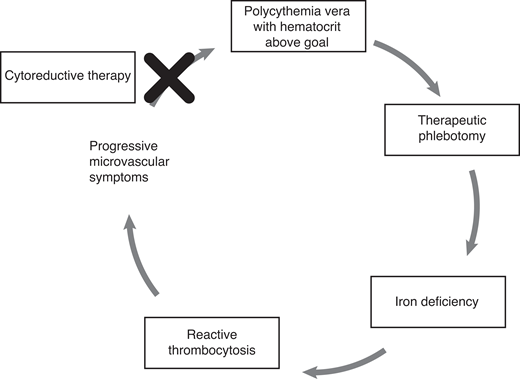
Comparative efficacy data to guide the selection of initial cytoreductive therapy in PV and ET are limited, and therapy is influenced by convenience, cost, tolerability, and safety. PegIFN is not currently approved in the US for treatment of ET or PV, yet consensus guidelines recommend its consideration as a first-line option for cytoreduction, particularly in younger patients and those who desire pregnancy.10
Efficacy of pegIFN
Hematologic response
In trials using standard formulations of recombinant IFN, hematologic responses were observed in about 80% of patients, who also reported a significant reduction in MPN-associated symptoms, including pruritus. More recent studies with longer-acting peg-IFN have reported a similar effect size. Two independent phase 2 studies using peginterferon alfa-2a in PV and ET both demonstrated effective cytoreduction with less discontinuation due to toxicity. The French “PV-Nord” group (PVN1) conducted a study of 37 patients with newly diagnosed PV and boasted count normalization in 95% of the cohort at 12 months.11 Testifying to the tolerability of this formulation, the vast majority of patients remained on the treatment for at least 1 year. Surprisingly, a great majority of these responses were durable at 6-year follow-up, even after stopping or switching therapies.12 In a separate study of patients with both PV and ET, response rates were around 80% for both disease subtypes.13 Noncomparative studies have defined the effectiveness of pegIFN in hydroxyurea (HU) resistance/intolerance.14 While these have both garnered support for their efficacy in normalizing blood cell counts and reducing phlebotomy needs, randomized trials comparing pegIFN to HU may also provide important direct comparative efficacy. MPN-RC-112 is a trial that randomizes high-risk PV and ET patients to first-line treatment with either pegIFN or HU. A final analysis of the trial has yet to be published, but interim trial updates report an overall response rate of 78% with peginterferon alfa-2a, not significantly different from that seen with HU (Table 1). Symptom benefit appears to be most pronounced for both agents in patients with a high baseline symptom burden, though more patient-reported outcome data are needed to make conclusions about symptom benefit for both HU and pegIFN. Comparative rates of thromboses will be a key end point for determining the best treatment across the age span.
Long-acting interferon trials
| Study | Phase | Comparison | Patients | Follow-up duration | Hematologic CR | Molecular response |
|---|---|---|---|---|---|---|
| PVN1 | 2 | peginterferon alfa-2a | 37 with PV | 31.4 mo | 94.6% | 18.9% undetected 72.4% with >50% reduction |
| MDACC | 2 | peginterferon alfa-2a | 40 with PV 39 with ET | 20 mo | 70% (PV) 76% (ET) | 14% and 6% undetected (PV vs ET) 54% and 38% with >50% reduction (PV vs ET) |
| PV-PROUD/CONTINUATION-PV | 3 | Ropeginterferon vs HU | 257 with PV | 45.5 mo | IFN vs HU 71% vs 51% at 36 months | 44% vs 51% at 12 mo (IFN vs HU) 66% vs 27% at 36 mo (IFN vs HU) |
| Study | Phase | Comparison | Patients | Follow-up duration | Hematologic CR | Molecular response |
|---|---|---|---|---|---|---|
| PVN1 | 2 | peginterferon alfa-2a | 37 with PV | 31.4 mo | 94.6% | 18.9% undetected 72.4% with >50% reduction |
| MDACC | 2 | peginterferon alfa-2a | 40 with PV 39 with ET | 20 mo | 70% (PV) 76% (ET) | 14% and 6% undetected (PV vs ET) 54% and 38% with >50% reduction (PV vs ET) |
| PV-PROUD/CONTINUATION-PV | 3 | Ropeginterferon vs HU | 257 with PV | 45.5 mo | IFN vs HU 71% vs 51% at 36 months | 44% vs 51% at 12 mo (IFN vs HU) 66% vs 27% at 36 mo (IFN vs HU) |
Molecular effects: defining disease modification
In addition to impressive hematologic responses, studies using pegIFN have reported patients achieving a complete molecular response. Defined as an undetectable JAK2V617F allele frequency, for the first time these data suggested that exogenous IFN could potentially be directly targeting the MPN clone.11 The JAK2V617F allele burden was progressively reduced in about 70% of PV patients and 40% of ET patients. Moreover, the JAK2V617F mutation became undetectable in 24% of PV patients in the PVN1 study.15 Slightly lower rates of molecular remission—15% in PV and 6% in ET patients—were reported in the US study. This is not surprising given a more heterogeneous study population with a longer disease duration. In both disease groups, the presence of additional mutations such as TET2, ASXL1, IDH2, and TP53 was associated with poorer molecular responses.16 Along with improvements in both symptoms and cell counts, these molecular responses raise the possibility and hope that this treatment could selectively and durably target the malignant clone and even allow hematologists to contemplate a curative treatment. This aspiration is often captured with the phrase “disease modifying” when treating patients with PV and ET.
Will I be on this forever? Treatment duration with pegIFN
Whether durable disease modification or cure is achievable with pegIFN has not yet been answered. How these generally encouraging results translate into clinical practice can be variable, and the optimal treatment duration with pegIFN is unknown. If a complete molecular remission is achieved, is a maintenance phase of treatment required? Tolerability and safety are critical to these decisions, particularly in a population with often near-normal life expectancy. Longer-term follow-up studies suggested an overall discontinuation rate of 22% for treatment-related toxicity. These rates decreased over time, though treatment-limiting grade 3 or 4 toxicities were still observed after 60 months on therapy.17 If provider and patient elect to pursue a treatment-free remission period in patients achieving a complete molecular response, what are the parameters for resuming treatment? Should blood count monitoring only drive retreatment decisions, or is monitoring for earlier measurable residual disease by JAK2V617F polymerase chain reaction a better standard? If the latter, what is the threshold of detectability driving decisions? At what frequency should patients be monitored? These and many more related questions remain unanswered.
Vascular risk
Many of the major life-limiting consequences of MPN stem from vascular complications, with rates of thrombosis reported at 5.5 events per 100 patients per year.18 The published longitudinal data suggest that IFN use may mitigate vascular risk, but comparative data are lacking to robustly answer these important questions. Encouragingly, the PVN1 trial did not report any thrombosis events in 6 years of follow-up, while the phase 2 US trial reported a thrombosis incidence of 1.22 per 100 patients per year.17 Additional long-term follow-up may help define any true vascular benefit, but these data suggest that pegIFN is associated with an overall low thrombosis risk.
Patients with splanchnic vein thrombosis (SVT) represent a particularly high-risk group. MPN are a common predisposing risk factor for SVT, involving hepatic, splenic, portal, or mesenteric veins, and it has been recognized that patients with SVT are at increased risk of developing recurrent thromboses, bleeding complications, and arterial thromboses. Furthermore, HU fails to prevent recurrent SVT.19 The MPD-RC 111 study set to address whether IFN could potentially fill this clinical need in a prospective trial evaluating peg-IFN in 20 patients with a history of SVT.14 Hematologic response rates were 70% at 12 months, on par with previous studies of all-comers, and SVT did not recur in any patients during 2.2 years of follow-up.
Ropeginterferon alfa-2b
Efforts to further improve the activity and tolerability of pegIFN treatment resulted in the development of ropeginterferon alfa-2b, a monopegylated IFN dosed every 14 days. The European Medicines Agency approved ropeginterferon alfa-2b in 2019 for PV without symptomatic splenomegaly; a decision from the US Food and Drug Association was expected in March 2021 but has yet to be decided at the time of this writing. If approved, ropeginterferon alfa-2b would be the first IFN specifically approved for MPN in the US. European Medicines Agency approval was based on a registration study known as PROUD-PV and its extension study, CONTINUATION-PV. These studies evaluated ropeginterferon alfa-2b compared with HU using a noninferiority design. PROUD-PV enrolled 257 PV patients with early-stage disease, including patients newly requiring cytoreductive therapy or those who had been treated with HU for fewer than 3 years.20 Randomization was stratified by age, history of thromboembolism, and prior HU use. The primary outcome was composed of hematologic response and normalization of spleen size at 12 months, achieved in 21% of patients in the ropeginterferon alfa-2b group and 28% in the HU group. Longer treatment and follow-up showed that 53% of patients achieved a complete hematological response with improved disease burden in the ropeginterferon alfa-2b group vs 38% of patients receiving HU (P = .044 at 36 months). Thromboembolic events in both arms were similarly rare. Reductions in JAK2V617F allele frequency appeared to improve over time, predominantly in the ropeginterferon alfa-2b group, supporting the disease-modifying claim that appears to be unique to pegIFN as a class.
Dosing: pegIFN and ropeginterferon
High doses of IFN are poorly tolerated. This was established in the nonpegylated formulation era and in dose-finding studies of pegIFN using up to 360 µg weekly.13 But determination of the optimal starting and maintenance dose for both pegIFN and ropeginterferon is a work in progress. Conventional pharmacodynamic studies are challenging when the target(s) and mechanism of action of these agents remain somewhat obscure. In addition, the relatively long time to response, with slow hematologic responses and subsequent deeper molecular responses, begs a philosophical dilemma between “hit it hard and hit it early” vs “slow and steady wins the race” since a linear and predictable dose-response and dose-toxicity relationship does not appear to exist, and maintaining patients on a tolerable dose for a long period of time may be of particular importance. Our approach is to start at a low dose, 45 µg weekly, of pegIFN alpha-2a (Pegasys, the only currently available US formulation) for a minimum of 4 weeks. Close monitoring of mood, liver, and thyroid function dictate further dose modifications or interruptions. Some patients stay at 45 µg weekly, and others increase to 90 µg weekly; doses as high as 135 or 180 µg weekly are less common but can also be tolerated by some and may be helpful in the absence of a hematologic response at lower doses.
Potential toxicities and monitoring for IFN therapies
Although longer-acting IFNs have improved tolerability compared to standard formulations, patients still commonly experience adverse effects. Fatigue and flu-like symptoms are prevalent. These tend to respond to acetaminophen or nonsteroidal anti-inflammatory medications and can abate with time. Efforts to minimize intolerabilities include low-dose run-in periods with dose-escalation algorithms. Toxicity monitoring during treatment includes blood counts as well as period thyroid and liver function testing and depression screening. Dose adjustments for cytopenias, liver dysfunction, and mood or neurocognitive effects are critical to safety and optimal response.
Are there differential responses to IFN depending on mutations?
As mentioned, a post hoc analysis of an early pegIFN study astutely observed that failing to achieve a complete molecular remission was associated with molecularly more complex disease beyond the driver mutation. The presence of mutations in TET2, ASXL1, EZH2, DNMT3A, and IDH1/2 decreased the likelihood of achieving a molecular remission.16 Much more recently, detailed analyses of serial samples found that leukemic progenitor cells with homozygous JAK2V617F mutations were more sensitive to IFN compared to cells with a single (heterozygous) JAK2V617F mutation and CALR-mutated cells. In addition, for the heterozygous JAK2V617F cells, a higher dose of IFN appeared to be important for effective clearance of the clone.21 Thus, response to IFN appears to be influenced by driver mutation type and zygosity and by additional disease-associated mutations.
Pregnancy
What will we tell our patient about her desire for additional pregnancies? While data on the management of PV in the setting of pregnancy are limited, the available studies indicate that early fetal loss is common, as is intrauterine growth restriction. Preterm delivery is common, and there is an estimated fetal survival of 50%.22 In general, the continuation of aspirin is recommended for virtually all pregnant women, and IFNs are considered safe and should generally be continued in patients for whom cytoreductive treatment was indicated prepregnancy, either for vascular risk reduction or symptomatic control.
CLINICAL CASE (continued)
Now 2 years old, our patient's daughter is thriving and meeting all her developmental milestones. Our patient started TP, continues on aspirin, transitioned to a non-estrogen-containing reliable form of contraception, and initiated pegIFN alpha-2a, initially at a low dose (45 µg weekly) and after 6 weeks, without discernable side effects, increased to 90 µg weekly. Her blood counts have improved, and her phlebotomy requirements have decreased. She is following with her obstetrics group and with maternal fetal medicine, planning for a second pregnancy. She will continue with both aspirin and pegIFN throughout pregnancy if she continues to tolerate it as well as prophylactic doses of low-molecular-weight heparin for 6 weeks postpartum.
Conclusions
Patients with PV and ET have a generally favorable prognosis, usually surviving decades and living full lives. But they live with an incurable chronic illness that for some has debilitating symptoms, and harder to capture is the existential uncertainty patients feel about the possibility of disease progression. There is excitement around pegIFNs, nontargeted therapies that selectively target the malignant MPN clone. Many foundational questions—who, when, for how long—require more precise answers, but the progress toward better outcomes is undeniable.
Conflict-of-interest disclosure
Elizabeth A. Traxler: no competing financial interests to declare.
Elizabeth O. Hexner: no competing financial interests to declare.
Off-label drug use
Elizabeth A. Traxler: off-label drug use includes hydroxyurea, peginterferon alfa-2a, and Ropeginterferon alfa-2b.
Elizabeth O. Hexner: off-label drug use includes hydroxyurea, peginterferon alfa-2a, and Ropeginterferon alfa-2b.
