

Abstract
The last 10 years have seen an explosion in the amount of data available through next-generation sequencing. These data are advancing quickly, and this pace makes it difficult for most practitioners to easily keep up with all of the new information. Complicating this understanding is sometimes conflicting information about variant pathogenicity or even about the role of some genes in the pathogenesis of disease. The more widespread clinical use of sequencing has expanded phenotypes, including the identification of mild phenotypes associated with previously serious disease, such as with some variants in RUNX1, MYH9, ITG2A, and others. Several organizations have taken up the task of cataloging and systematically evaluating genes and variants using a standardized approach and making the data publicly available so that others can benefit from their gene/variant curation. The efforts in testing for hereditary hemorrhagic, thrombotic, and platelet disorders have been led by the International Society on Thrombosis and Haemostasis Scientific Standardization Committee on Genomics in Thrombosis and Hemostasis, the American Society of Hematology, and the National Institutes of Health National Human Genome Research Institute Clinical Genome Resource. This article outlines current efforts to improve the interpretation of genetic testing and the role of standardizing and disseminating information. By assessing the strength of gene–disease associations, standardizing variant curation guidelines, sharing genomic data among expert members, and incorporating data from existing disease databases, the number of variants of uncertain significance will decrease, thereby improving the value of genetic testing as a diagnostic tool.
Learning Objectives
Define the current resources available to clinicians, researchers, and patients to allow data sharing and standardization
Identify the current ongoing efforts to improve and standardize interpretation and allow categorization of variants of uncertain significance
Understand the role of the Clinical Genome Resource and ClinVar in standardizing and disseminating gene-specific variant curation results
Clinical case
A 14-year-old girl presented to a hematology clinic for evaluation of menorrhagia and long-standing thrombocytopenia. She had initially been noted to have thrombocytopenia at about 15 months of age, shortly after starting to walk, when she was presented to her primary care physician with bruising. At that time, she was also noted to have some petechiae. A complete blood count demonstrated normal counts, except for a platelet count of 55 × 109/L. Evaluation at that time provided a diagnosis of immune thrombocytopenia. She was treated with intravenous immunoglobulin and prednisolone, neither of which increased her platelet count above 50 to 60 × 109/L. She had bleeding symptoms of minor epistaxis as well as bruising and petechiae but was otherwise well, and no further attempts to alter platelet counts were made. At 14 years of age, she experienced menarche, which resulted in hemorrhage to a hemoglobin of 5.4 g/dL over 10 days with heavy menstrual bleeding. She was admitted, transfused with packed red blood cells, and treated again with intravenous immunoglobulin and prednisone as well as a platelet transfusion for a platelet count at that time of 37 × 109/L. Review of her peripheral smear demonstrated >50% large or giant platelets. She was then transfused with platelets, and a 45-minute post-platelet count demonstrated an appropriate rise, with her platelet count 24 hours later still being >100 × 109/L. She was referred to hematology for further evaluation of her bleeding and thrombocytopenia. Evaluation at that time was concerning for inherited platelet disorder, despite lack of family history of thrombocytopenia or associated syndromes. A genetic evaluation demonstrated 2 variants in the GP9 gene, inherited in trans with a maternally inherited known pathologic variant and de novo variant of uncertain significance (VUS).
Introduction
Rare diseases, defined as those affecting fewer than 1 in 2000 individuals in the general population, often have genetic causes, disproportionately affect children (50% to 75%), and tend to result in severe, multisystem disorders. Accurate genetic diagnosis of a rare disease enables access to disorder-specific support groups and provides information guiding management, therapy, and prognosis as well as determination of risk for family members.1,2 This makes appropriate and accurate diagnosis important for many reasons. More recently, increased clinical sequencing has expanded phenotypes and revealed that some rare diseases are more common than originally thought and not always associated with severe phenotypes.3 Among the platelet disorders with expanding phenotypes are RUNX1-related disorder,4 Bernard-Soulier syndrome,5 and others.6 Even with Glanzmann thrombasthenia, some individuals are not diagnosed until later in life.7
Genetic testing started out with low-resolution cytogenetic testing, moving to microarray8 and then the development of multiplexed single-gene sequencing in panels and then eventually high-throughput next-generation sequencing (NGS) of whole exomes and then whole genomes.9-12 There are advantages of NGS testing, allowing massively parallel sequencing of millions to billions of genetic loci simultaneously, but this may come at the price of loss of resolution for some areas of the genome and inability to detect some types of copy number variation, which may have important implications, particularly in some types of disorders, such as those caused by small deletions (thrombocytopenia-absent radii syndrome),13 or due to changes in genes with associated pseudogenes or to variations in copy number repeat changes (Quebec platelet syndrome).14 One of the difficulties in NGS is the appropriate determination of which variants are clinically significant.12,15 Sequencing so many genetic loci may provide thousands of single-nucleotide variants in a single individual, leaving the laboratory with the difficult task of assessing the clinical relevance of all of these genetic changes. Several groups have worked to develop tools and consensus to allow harmonization of clinical validity, pathogenicity, and clinical utility of genetic variants.16
Process of genetic test interpretation
When a patient is referred for genetic testing, several key elements should be in place to ensure accurate and timely testing as well as appropriate interpretation and return of results.
Test selection
The clinician ordering the test needs to determine the appropriate test to send on the basis of clinical phenotype and the suspected diagnosis. Various testing strategies exist and can be employed to arrive at a molecular diagnosis, including sequencing of targeted suspected genes, sequencing a panel of genes related to a specific disease or phenotype, or whole-exome or whole-genome sequencing, which is more agnostic to the diagnosis or potential genes involved.17 Clinicians ordering genetic testing must be familiar with the available testing and be able to order the appropriate test for the disease category (Table 1). For example, patients with severe factor VIII deficiency rarely require exome sequencing to arrive at a molecular diagnosis, but instead targeted sequencing including examination for small deletions and duplications is appropriate in evaluating a patient with hemophilia due to factor VIII deficiency. In contrast, individuals with platelet function defects benefit from careful functional evaluation and, if appropriate, may benefit from broader panels or even exome sequencing to molecularly define the etiology of their disorder.
Types of genetic testing and their advantages and disadvantages
| Test type | Advantages | Disadvantages | Example of best use |
|---|---|---|---|
| Sanger sequencing (single gene) | • Less expensive | • Only provides information on one gene | Confirmation of known variant in affected family |
| • Fast turnaround | |||
| Gene panel | • More in-depth coverage of genes of interest | • Does not interrogate genes outside of panel (may miss novel associations) | Clinical scenario provides suspected diagnosis for confirmation |
| • Custom designed to disease of interest | • Utility depends on frequency of panel update | ||
| • No secondary findings | |||
| • Generally faster turnaround | |||
| Whole exome | • Agnostic and comprehensive | • Insensitive to deletions/duplications, depending on technique | Novel syndrome unclear diagnosis |
| • Allows identification of novel variants | • Reading depth influences ability to find variants in some genes | ||
| • May be more readily available (not custom) | • Expensive | ||
| Whole genome | • Allows sequencing of noncoding regions | • Expensive | Suspected genetic disease due to noncoding variant |
| • Better than WES at detecting indels/duplications/inversions | • Lower read depth for some areas | ||
| CGH array | • Detects large genomic changes | • Insensitive to single-nucleotide changes | Chromosomal deletions, loss |
| Test type | Advantages | Disadvantages | Example of best use |
|---|---|---|---|
| Sanger sequencing (single gene) | • Less expensive | • Only provides information on one gene | Confirmation of known variant in affected family |
| • Fast turnaround | |||
| Gene panel | • More in-depth coverage of genes of interest | • Does not interrogate genes outside of panel (may miss novel associations) | Clinical scenario provides suspected diagnosis for confirmation |
| • Custom designed to disease of interest | • Utility depends on frequency of panel update | ||
| • No secondary findings | |||
| • Generally faster turnaround | |||
| Whole exome | • Agnostic and comprehensive | • Insensitive to deletions/duplications, depending on technique | Novel syndrome unclear diagnosis |
| • Allows identification of novel variants | • Reading depth influences ability to find variants in some genes | ||
| • May be more readily available (not custom) | • Expensive | ||
| Whole genome | • Allows sequencing of noncoding regions | • Expensive | Suspected genetic disease due to noncoding variant |
| • Better than WES at detecting indels/duplications/inversions | • Lower read depth for some areas | ||
| CGH array | • Detects large genomic changes | • Insensitive to single-nucleotide changes | Chromosomal deletions, loss |
CGH, comparative genomic hybridization; WES, whole-exome sequencing.
Many centers make available genetic counselors to aid in the appropriate selection of testing and subsequent consent and counseling of patients around the test and results (see below). The use of genetic counselors, if available, is recommended and provides an additional perspective both on the need for testing and on the potential risks/benefits of outcomes.18 In addition, consultation with a disease expert may be helpful to determine the appropriate test and the best place to send the test among the various options to ensure timely results and aid with interpretation once those results are available.
Consent for testing
Once the appropriate genetic test has been identified, a clear conversation with the patient and, if appropriate, the patient’s family should ensue with discussion of the risks and benefits of genetic testing. Particular ethical considerations may play a role in pediatric patients19 who are more likely to carry genetic diagnoses but also may not be capable yet of participating in decision making about implications of results of genetic diagnoses. Full disclosure of not only the potential benefits of genetic testing but also the risks is critical to appropriate consent. Families who will be undergoing less targeted sequencing (panels or exome sequencing) need to understand risks of return of cancer predisposition, as well as incidental and secondary findings, and should be given the opportunity to “opt out” of especially some types of findings.20 The consent and decisions about return of secondary findings should be documented in the medical record. The American College of Medical Genetics (ACMG) has published a list of secondary findings that are considered “medically actionable”21 and recommends return of these results because these are results regarding which standard clinical interventions exist to prevent or mitigate the disease. Most participants in clinical studies have indicated a preference for return of secondary findings,22-24 but these may not fully represent all patients (eg, those who would choose not to participate in research). Finally, regulations vary in some countries and should also be considered when providing consent because they may alter the availability of testing.
Interpretation and return of results
A patient’s genetic data are analyzed, and each variant is assigned a classification based on the available literature and, today, information in public databases that helps to gather genetic testing information to inform the pathogenicity of findings. Variants may be “called” in 1 of 5 categories (Figure 1). Generally, variants that are benign or likely benign are not reported in genetic testing reports, because these are variants that have reliably been shown to be tolerated sequence variations. Definitive diagnostic results are returned with pathologic or likely pathologic variants identified in genes associated with the described phenotype. The result of a VUS needs to be carefully considered but does not mean the patient carries the associated diagnosis, and some VUSs have, since their initial description, been defined as benign. Return of a VUS may require consultation with genetic counselors, disease experts or additional testing and additional follow-up. If the results of testing are inconclusive, follow-up testing/analysis in 2 to 3 years may be advised because genetic testing is evolving with continued identification of additional genes and variants.
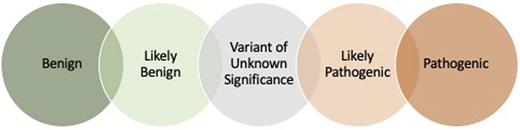
Variant classifications. On the basis of American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines,34 the variant classifications have been proposed along with a scoring system to standardize variant interpretation across clinical laboratories.
Variant classifications. On the basis of American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines,34 the variant classifications have been proposed along with a scoring system to standardize variant interpretation across clinical laboratories.
Finally, once the results of a genetic test are known, those results must be returned to the patient, which involves a complete discussion of the findings within the context of that patient’s disease. Patients who carry single variants for autosomal recessive disorders generally do not have symptoms (although some genes are now being recognized as much more gene dose dependent than previously appreciated). Appropriate disclosure and discussion of the clinical significance of a VUS is one of the biggest challenges in genetic diagnosis, and several efforts are ongoing to help move variants out of this area of uncertainty.
Challenge of VUS
A VUS, generally a variant that has not previously been reported, cannot be conclusively assigned to either a benign or pathologic category on the basis of a single patient. Even if the variant has previously been reported, there may be conflicting information about how that variant may affect the gene product or even about the validity of the gene association.25 Aggregation of patients with similar phenotypes/variants is a powerful tool to help sort VUSs into more helpful variant classifications.26-28 In addition, experimental validation of the detrimental effect of a variant on protein function or expression can also help to define the role of a particular variant in the molecular pathogenesis of disease.29
Databases that collate and curate the vast amounts of information, making it available to other laboratories in interpreting results, allow more streamlined interpretation and better care for patients. Several databases have arisen over time; some are disease specific, collecting information about RUNX1 variants, Glanzmann thrombasthenia–associated variants, and Bernard-Soulier syndrome–associated variants, whereas others are more general, collating information about any variant in any gene.
Development of public resources to assist in variant calling
There are multiple groups that have contributed to growing knowledge, especially within the genes responsible for inherited platelet disorders, including ThromboGenomics (http://thrombo.cambridgednadiagnosis.org.uk), the UK Genotyping and Phenotyping of Platelets group,30 the Biomedical Research Centres/Units Inherited Diseases Genetic Evaluation Bleeding and Platelet Diseases consortium,31 and groups in Spain32 and Denmark.33 These groups have pioneered the development of gene panels and NGS techniques for research and clinical purposes. Although a full discussion of the individual contributions of each of these groups, or even of the ways in which this field has advanced, is beyond the scope of this discussion, with their work came the explosion in information requiring curation and a need for a standardized approach to variant calling. As a direct result of international collaborations, several public resources have been developed over the past decade to help centralize information and provide open resources to scientists, clinicians, laboratories, and the public. Therefore, examining some of these resources and their roles and goals in the context of genetic testing may help in understanding how they can improve testing and serve as a model for comprehending the broader role of public resources in standardization.
ClinVar: a public database of the National Center for Biotechnology Information at the National Institutes of Health
ClinVar was launched in 201316 at the National Center for Biotechnology Information at the National Institutes of Health to provide a database of genetic variants that is centralized and open access to aid in interpretation of variants. One of the key missions of ClinVar was to gather the data generated from clinical genetic testing (variants and interpretations) and to add this together with other information about variants, providing a single source of information. The focus was for clinical laboratories to share variants, interpretations, and evidence for interpretations to improve variant interpretation overall. ClinVar then partnered with the Clinical Genome Resource (ClinGen) to create expert panels to provide the US Food and Drug Administration–approved database with consensus panel interpretation of all of the amassed data within ClinVar. ClinVar provides information within the database about whether variants have undergone any review and the level of that review (from single submission to aggregate interpretation to expert panel review). The level of evidence is provided and given a star rating from 0 to 4 stars, with 0 stars for a variant from a single submitter with criteria supporting an assertion and 4 stars representing evidence-based practice guidelines (eg, ACMG, Clinical Pharmacogenetics Implementation Consortium). The expert panel curations are given 3 stars, and there are multiple expert panels within ClinGen and recognized outside panels, including Pharmacogenomics Knowledgebase, Evidence-based Network for the Interpretation of Germline Mutant Alleles, and the panel for CFTR2.
ClinGen
ClinGen (https://clinicalgenome.org/), in contrast, was established to create evidence-based consensus for curating genes and variants, develop machine learning strategies, and disseminate collective knowledge and resources for unrestricted community use. ClinGen incorporates ClinVar and other resources (including extensive use of expert panels, some of which have been convened by the American Society of Hematology) to ask several questions (eg, Is the gene associated with disease? Is the variant causative? Is the information actionable?) about genes and variants and gather available knowledge to reach consensus.
ClinGen brings together physicians, clinical geneticists, laboratorians, research scientists, and biocurators to develop standards for variant and gene interpretation and dissemination of information. The work is split into several clinical domain working groups (CDWGs), and within these CDWGs, there are variant curation expert panels that specify the rules for variant curation (ie, How does one establish that a variant is pathologic?) and then curate genes/gene families as an expert panel. In addition, ClinGen has several gene–disease curation expert panels that curate the strength of association between a particular gene and phenotype/disease. ClinGen also has multiple other working groups/areas dedicated to improving variant interpretation guidelines, community curation, complex disease associations, somatic variability, and determining the actionability of secondary findings.
Hemostasis/Thrombosis CDWG: American Society of Hematology–ClinGen–ThromboGenomics partnership
In 2018, the Hemostasis/Thrombosis CDWG (https://www.clinicalgenome.org/working-groups/clinical-domain/hemostasis-thrombosis/) was established to improve the utility of genetic testing within this clinical domain. On the basis of prior experience of the ThromboGenomics group and the International Society on Thrombosis and Haemostasis Scientific Standardization Committee (ISTH-SSC) on Genomics in Thrombosis and Hemostasis, this international group of experts was organized to work in partnership to curate genes and variants within multiple domains of thrombosis and hemostasis. These curation results are publicly available on the ClinGen website (https://clinicalgenome.org/). The Hemostasis/Thrombosis CDWG within ClinGen has established several expert curation panels, as well as a gene–disease curation panel for this domain (Figure 2).
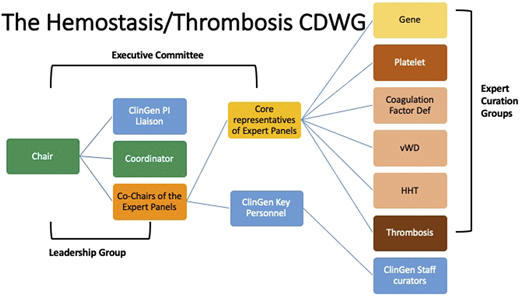
The expert panels within this CDWG have, over the past 2 years, curated 42 gene–disease associations and have established 5 variant curation expert panels whose variant curation rules are in various stages of approval by ClinGen. In partnership with ISTH-SSC, the gene curation panel was able to get a head start on platelet disorder gene curation by starting with the list of hemostasis genes that had been assigned a high likelihood of pathogenicity (https://www.isth.org/page/GinTh_GeneLists). They then harmonized the gene–disease associations of the 2 groups, providing a curated list of genes that had already been reviewed by the expert panel. This ongoing partnership benefits from the yearly platelet disorder gene review at the ISTH meeting and allows rapid incorporation of newly described genes into the list of associated genes.
Resolution of the case
The patient returned to the office to discuss the findings of her genetic testing. After discussion, her family opted for functional and flow cytometric evaluation to confirm the clinical diagnosis of Bernard-Soulier syndrome. Flow cytometry demonstrated markedly reduced expression of glycoprotein 1b/IX receptor on platelet surfaces, and platelet aggregation studies were limited due to thrombocytopenia but did demonstrate essentially normal aggregation to adenosine 5′-diphosphate and collagen and absent agglutination with ristocetin. The patient and family agreed to submission of the additional functional information to the laboratory to better define the VUS. Subsequently, the laboratory used this information, along with the clinical features and follow-up experimental evidence, for a submission to ClinVar through the online submission portal. Consent for submission was documented in the medical record along with the original consent for genetic testing.
The diagnosis of Bernard-Soulier syndrome markedly altered ongoing management, changing the discussions about the underlying pathophysiology of the disease, best available treatments, and risk of bleeding as well as conversations of about heritability and risks to any future siblings or offspring.
Future directions and ongoing needs
The development of these public databases, which have benefited from the work of prior public resources, has the potential to markedly improve genetic diagnoses by allowing the community to more quickly define the role of novel genes and variants in disease. Continued collaboration between researchers, clinical laboratories, and clinicians in sharing variants (regardless of the status) along with descriptive information allows development of more robust data and ultimately benefits the entire community.
Acknowledgments
The author acknowledges the work of the Hemostasis/Thrombosis Clinical Domain Working Group, on behalf of whom this article is presented. Especially, she acknowledges Kristy Lee, the program coordinator, and Nigel Key, the committee chair, for their leadership and oversight.
Correspondence
Michele P. Lambert, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd., Abramson Research Center, Room 316C, Philadelphia, PA 19104; e-mail: lambertm@email.chop.edu.
