

Abstract
As CAR T-cell therapy has advanced in B-cell acute lymphoblastic leukemia, research is now underway to develop similar therapies for other lymphoid and myeloid malignancies for pediatric patients. Barriers, including antigen selection and on-target/off-tumor toxicity, have prevented the rapid development of immune-based therapies for T-lineage and myeloid malignancies. More recently, unique strategies have been developed to overcome these barriers, with several products advancing to clinical trials. For T-lineage diseases, targets have focused on CD5, CD7, and CD38, whereas myeloid disease targets have predominately focused on CD123, CD33, and, more recently, CLL-1. This review provides a comprehensive overview of these targets and approaches to overcoming safety concerns in the development of CAR T-cell therapies for pediatric patients with T-lineage and myeloid malignancies.
Learning Objectives
Recognize the barriers to CAR T-cell therapy for children with T-cell or myeloid hematologic malignancies
Understand strategies to overcome these barriers, including a review of current studies of CAR T cells for children with T-cell or myeloid malignancies
Introduction
Chimeric antigen receptor (CAR) T cells redirected against B-cell antigens (eg, CD19, CD22) have demonstrated remarkable clinical activity in children and adults with relapsed/refractory B-cell malignancies.1 Successful development of CAR T cells for non–B-cell hematologic malignancies has been far more challenging, primarily due to antigen selection and on-target/off-tumor toxicity.
Early efforts in non–B-cell hematologic malignancies focused on finding near-universal targets with permissible on-target/off-tumor toxicity, analogous to CD19. As the field has evolved, it has become apparent that such targets are unlikely to exist; instead, we must come up with more sophisticated strategies to support the implementation of CAR T cells in these diseases. Using a patient case scenario, we review the major obstacles to successfully implementing CAR T-cell strategies for two hematologic malignances in pediatrics with the highest unmet needs: acute myeloid leukemia (AML) and T-cell acute lymphoblastic leukemia and lymphoblastic lymphoma (T-ALL/T-LL). We then present strategies investigators are implementing to overcome these obstacles and review recent and current clinical trials.
Clinical case
A 3-year-old boy presented to a clinic with a large mediastinal mass and was diagnosed with T-LL. Flow cytometric immunophenotyping confirmed a diagnosis of T-LL on the basis of the presence of CD7, CD2, CD38, and cytoplasmic CD3. At the end of induction, his mediastinal mass had decreased in size, but following consolidation chemotherapy, his disease had progressed. After two salvage attempts, he continued to have disease progression as well as the development of new sites of disease. On the basis of his unresponsiveness to chemotherapies, his treating team began investigating immunotherapeutic options.
CAR T-cell therapy for children with T-ALL and T-LL
T-ALL is a more aggressive and more chemoresistant disease than B-cell acute lymphoblastic leukemia (B-ALL).2 Although outcomes for upfront disease have neared those for B-ALL, relapsed and/or refractory T-ALL is particularly difficult to treat and has dismal outcomes.3 Survival after relapsed/refractory T-LL is equally poor, with an overall survival of <10%.4 The poor prognoses associated with these diseases are compounded by a lack of immunotherapeutic salvage options. Barriers to the use of CAR T cells for patients with T-cell malignancies include product contamination with malignant T lymphoblasts, fratricide, and on-target/off tumor toxicity and the associated risk for T-cell aplasia (Figures 1 and 2).
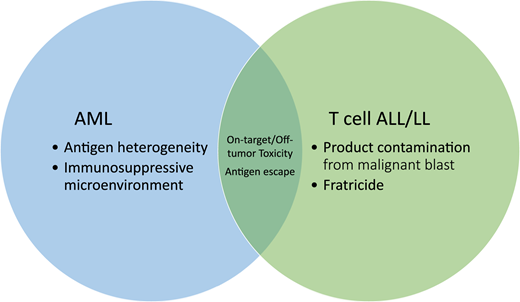
An overview of the barriers facing chimeric antigen receptor T-cell development for acute myeloid leukemia (AML) and T-cell acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LL).
An overview of the barriers facing chimeric antigen receptor T-cell development for acute myeloid leukemia (AML) and T-cell acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LL).
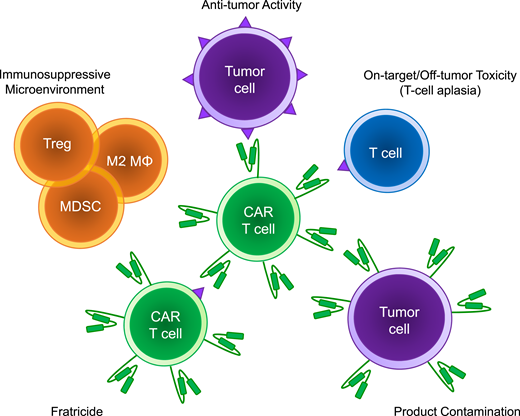
Schema of the barriers to implementation of chimeric antigen receptor T cells for non–B-cell hematologic malignancies. Treg, T regulatory cells; M2 MΦ, M2 macrophage; MDSC, myeloid-derived suppressor cell.
Schema of the barriers to implementation of chimeric antigen receptor T cells for non–B-cell hematologic malignancies. Treg, T regulatory cells; M2 MΦ, M2 macrophage; MDSC, myeloid-derived suppressor cell.
There are two issues unique to the manufacturing of a CAR T-cell product for T-cell disease: contamination with malignant cells and fratricide. Contamination of a CAR T-cell product with malignant cells is a theoretical risk for any patient with a hematologic malignancy. The implications of product contamination were highlighted by a recent case report of a patient with B-ALL who developed a CD19-negative relapse 8 months after a CD19 CAR T-cell infusion that was determined to have been from a leukemic cell that was transduced during manufacturing of the CAR product.5 This risk can be mitigated when the product undergoes upfront T-cell selection.6 However, the ability to select for T lymphocytes in patients with T-cell malignancies is more technically difficult because of the higher likelihood for circulating tumor cells and the shared antigen expression between malignant and normal T cells. Surface CD3 is commonly absent on T-ALL; thus, with meticulous separation techniques, contamination can be greatly reduced but not eliminated.
One proposed solution for preventing product contamination is the use of allogeneic, or “off-the-shelf,” T cells from healthy donors. Along with an inherent lack of risk for product contamination, this strategy has the additional benefit of being readily available, which is critically important for patients with rapidly progressing disease or those unfit to undergo apheresis. The major limitation of this approach is the risk for significant graft-versus-host disease (GVHD) without a full allogeneic match. Alternatively, a host immune system may reject a mismatched product, preventing engraftment. To eliminate the risk of GVHD, several groups are using gene-editing technology to knock out the T-cell receptor (TCR) α-chain. By knocking out this component, TCR-mediated signaling is blocked and GVHD is prevented while preserving the function of the CAR T cell.7 In the current state, the efficacy of allogeneic CAR T-cell products has lagged behind autologous products for treatment of B-cell malignancies.8,9
Natural killer (NK) cells are being explored as an alternative off-the-shelf product, with early clinical trial results in B-cell malignancies rivaling the outcomes of autologous CAR T cells, albeit in a small number of patients.10 There are numerous potential advantages to using NK cells over CAR T cells. Owing to their lack of a TCR, NK cells do not pose a risk for GVHD and therefore require no additional gene editing to be used as a universal product. Theoretically, NK cells may retain lytic activity against the tumor cells in a non–antigen-dependent manner, which may prove useful in settings where antigen modulation is frequently encountered. In addition, there is less antigen overlap between NK cells and non–B-cell hematologic malignancies, which may allow a greater number of possible targets.
Fratricide is a unique form of ex vivo on-target/off-tumor toxicity commonly referred to as “self-killing.” Fratricide is when CAR T cells attack each other during the manufacturing process and occurs if the target antigen is concurrently expressed on the CAR T cell (Figures 1 and 2). This is a major theoretical barrier preventing the targeting of T-cell malignancies because there is an inherent overlap of surface antigens between malignant and CAR T cells. Without alteration of the target antigen on the CAR T cells, CAR-mediated self-killing may occur, undermining T-cell expansion during the manufacturing phase. Using NK cells may be advantageous in avoiding fratricide when the T-cell antigen is not shared with the NK cells.
Aside from a move toward NK cells, a straightforward solution to avoid fratricide is choosing a tumor antigen that is highly specific for the malignant cells and not expressed on normal T cells. Unfortunately, the number of these potential targets is limited, and they are usually restricted to a subset of patients with T-ALL (Table 1). One example is CD1a, which, in the normal state, is an antigen largely restricted to developing cortical thymocytes. CD1a is also expressed in cortical T-ALL, a major subset of T-ALL in adult and pediatric patients, making it relatively tumor specific. Preclinical modeling has shown an absence of fratricide, and, notably, the CAR T cells appear to respond to viral antigens, suggesting they retain some anti-infectious function, reducing the concern for opportunistic infections.11
Currently identified T-cell antigens for immunotherapeutic targeting under preclinical or clinical study
| Target | T-ALL/T-LL frequency | Normal tissue expression | Selected references |
|---|---|---|---|
| CD1a | 80%/67% | Cortical thymocytes, immature dendritic cells, Langerhans cells | 11 |
| CD3 | 33%/46% | Thymocytes, mature T cells, NK cells | 40 |
| CD4 | 82%/72% | T-helper cells, thymocytes, granulocytes, monocytes, dendritic cells, Langerhans cells | 23 |
| CD5 | 88%/95% | Mature T cells, thymocytes, B cells | 17 |
| CD7 | 98%/97% | Mature T cells, thymocytes, NK cells | 20 |
| CD38 | 100%/100% | Plasma cells, hematopoietic progenitor cells, T cells, NK cells, B cells | 12 |
| CD123 | 43%/— | Plasmacytic dendritic cells, eosinophils | 41 |
| TRBC1 | 30%/— | T cells | 24 |
| Target | T-ALL/T-LL frequency | Normal tissue expression | Selected references |
|---|---|---|---|
| CD1a | 80%/67% | Cortical thymocytes, immature dendritic cells, Langerhans cells | 11 |
| CD3 | 33%/46% | Thymocytes, mature T cells, NK cells | 40 |
| CD4 | 82%/72% | T-helper cells, thymocytes, granulocytes, monocytes, dendritic cells, Langerhans cells | 23 |
| CD5 | 88%/95% | Mature T cells, thymocytes, B cells | 17 |
| CD7 | 98%/97% | Mature T cells, thymocytes, NK cells | 20 |
| CD38 | 100%/100% | Plasma cells, hematopoietic progenitor cells, T cells, NK cells, B cells | 12 |
| CD123 | 43%/— | Plasmacytic dendritic cells, eosinophils | 41 |
| TRBC1 | 30%/— | T cells | 24 |
NK, natural killer; T-ALL, T-cell acute lymphoblastic leukemia; T-LL, T-cell lymphoblastic lymphoma.
Another example of an antigen with relative tumor specificity is CD38.12 Targeting CD38 has proved to be safe and well tolerated in multiple myeloma, with some supporting preclinical and emerging clinical evidence in T-ALL.13,14 Along with restricted expression on normal T cells, additional attractive properties of CD38 are its plasticity and its ability to be upregulated in a variety of hematologic malignancies, enhancing the cytolytic activity of CAR T cells in preclinical studies.15 Related to the tolerability of other CD38-targeting strategies in multiple myeloma, clinical investigations involving CD38-targeting CAR T cells are quickly advancing.16
Strategies to avoid fratricide are required when a tumor-specific antigen cannot be identified and may involve indirect or direct modulation of universally expressed T-cell antigens on the CAR T cell. An example of indirect modulation is modeled through the targeting of CD5. After transduction of CAR T cells with a CD28 costimulatory domain, CD5 is downregulated either by internalization or by masking of the CD5 antibody epitope by the CAR. This downregulation limits the degree of fratricide in this product while preserving the ability of the CAR to target malignant cells. This product has been advanced to clinical trials, with some encouraging preliminary results available (Table 2).17,18 In contrast, when a 4-1BB costimulatory domain was incorporated, significant fratricide occurred, suggesting different costimulatory molecules affect antigen stabilization.19
Active chimeric antigen receptor T-cell clinical trials for T-cell acute lymphoblastic leukemia/T-cell lymphoblastic lymphoma
| Target | ClinicalTrials.gov identifier | Institution/sponsor | Age restriction | Status | Reference |
|---|---|---|---|---|---|
| CD4 | NCT03829540 | Stony Brook University | 18+ y | Recruiting | 23 |
| CD5 | NCT03081910 | Baylor College of Medicine | <75 y | Recruiting | 18 |
| CD7 | NCT04033302NCT04004637NCT03690011NCT04264078 | Shenzhen Geno-Immune Medical Institute | 6 mo-75 y | Recruiting | — |
| First Affiliated Hospital of Zhengzhou University | 7-70 y | Recruiting | 21 | ||
| Baylor College of Medicine | <75 y | Not yet recruiting | 7 | ||
| Xinqiao Hospital of Chongqing | 2-70 y | Not yet recruiting | — |
| Target | ClinicalTrials.gov identifier | Institution/sponsor | Age restriction | Status | Reference |
|---|---|---|---|---|---|
| CD4 | NCT03829540 | Stony Brook University | 18+ y | Recruiting | 23 |
| CD5 | NCT03081910 | Baylor College of Medicine | <75 y | Recruiting | 18 |
| CD7 | NCT04033302NCT04004637NCT03690011NCT04264078 | Shenzhen Geno-Immune Medical Institute | 6 mo-75 y | Recruiting | — |
| First Affiliated Hospital of Zhengzhou University | 7-70 y | Recruiting | 21 | ||
| Baylor College of Medicine | <75 y | Not yet recruiting | 7 | ||
| Xinqiao Hospital of Chongqing | 2-70 y | Not yet recruiting | — |
Summary of active and completed clinical trials according to www.clinicaltrials.gov as of 1 June 2020.
Alternatively, forced inhibition of the target antigen can be engineered in order to prevent fratricide. Examples of direct antigen modulation include gene editing or protein expression blockers (PEBLs); both are currently being implemented in CD7-targeting strategies. Early studies involving CD7 as a CAR target showed extensive fratricide because CD7 is not downregulated in a fashion similar to CD5. By using a CRISPR/Cas9 system, CD7 can be knocked out, eliminating surface expression and the risk of fratricide, with some of these products advancing to clinical trials (Table 2).7,20,21 Similarly, a PEBL couples an anti-CD7 single-chain variable fragment with an endoplasmic reticulum/Golgi-retention motif, preventing any newly synthesized CD7 from being expressed. This technique was remarkably effective at abrogating CD7 expression and eliminating fratricide without compromising CAR T-cell function in a preclinical model.22 An additional advantage to the PEBL approach is that it does not require additional gene editing to the T cells.
A final primary concern, discussed in more detail below, is the concept of on-target/off-tumor toxicity and the risk of targeting healthy T cells in vivo, leading to T-cell aplasia (Figures 1 and 2). Strategic antigens that may be suitable for circumventing prolonged T-cell aplasia include CD4 and/or TRBC1/223,24 (Table 1). These antigens are expressed in a subset of normal T cells, but, given the clonal nature of malignancies, they are universally expressed on the tumor in a subset of patients. By targeting these antigens, there would be a significant reduction in a patient’s normal T-cell compartment but potentially sufficient T-cell populations to limit life-threatening infections if only transiently eliminated.
CAR T-cell therapy for children with AML
Relapsed and/or refractory AML remains a major source of childhood cancer–associated mortality.25 Similarly, treatment-related leukemia, including lineage switch after immunotherapeutic pressures, is particularly difficult to salvage.26 These challenging diseases have prompted an interest in novel immunotherapeutic strategies for myeloid diseases.
The major challenge in the treatment of AML is antigen selection. This is due in part to the phenotypic heterogeneity not only between patients but also within an individual patient, limiting the identification of a universal target (Table 3). When choosing an antigen with restricted expression, there is a risk of incompletely treating the tumor or precipitating an antigen escape. On the opposite end of the spectrum is choosing a target too widely expressed, leading to substantial on-target/off-tumor toxicity (Figures 1 and 2).
Currently identified acute myeloid leukemia antigens for immunotherapeutic targeting under preclinical or clinical study
| Target | Frequency in AML | Normal tissue expression | Selected references |
|---|---|---|---|
| CD7 | 10%-35% | Mature T cells, thymocytes, NK cells | 42 |
| CD13 | 75%-95% | Granulocytes, monocytes, mast cells, osteoclasts | 43 |
| CD33 | 75%-95% | Hematopoietic progenitor cells, basophils, granulocytes, mast cells, monocytes, Kupffer cells | 42 |
| CD38 | 43% | Plasma cells, hematopoietic progenitor cells, T cells, NK cells, B cells | 12 |
| CD44v6 | 64% | Keratinocytes | 44 |
| CD70 | 95% | B cells, T cells, dendritic cells | 45 |
| FLT3 (CD135) | 50% | Hematopoietic stem cells, neurons, testis | 46 |
| CD123 | 97% | Myeloid progenitors, plasmacytic dendritic cells, eosinophils, endothelial cells | 42 |
| Lewis Y (CD174) | 46% | Hematopoietic progenitor cells, intestinal epithelial cells | 47 |
| CD244 | 95% | NK cells, T cells, monocytes, basophils, eosinophils, spleen | 42 |
| TIM3 (CD366) | 85% | T cells, lung tissue | 42 |
| CLL-1 (CLEC12A, CD371) | 80% | Granulocytes, monocytes | 30,31 |
| Folate receptor B | 70% | Myeloid cells | 48 |
| NKG2DL | 70% | T-regulatory cells, endothelial cells | 49 |
| Target | Frequency in AML | Normal tissue expression | Selected references |
|---|---|---|---|
| CD7 | 10%-35% | Mature T cells, thymocytes, NK cells | 42 |
| CD13 | 75%-95% | Granulocytes, monocytes, mast cells, osteoclasts | 43 |
| CD33 | 75%-95% | Hematopoietic progenitor cells, basophils, granulocytes, mast cells, monocytes, Kupffer cells | 42 |
| CD38 | 43% | Plasma cells, hematopoietic progenitor cells, T cells, NK cells, B cells | 12 |
| CD44v6 | 64% | Keratinocytes | 44 |
| CD70 | 95% | B cells, T cells, dendritic cells | 45 |
| FLT3 (CD135) | 50% | Hematopoietic stem cells, neurons, testis | 46 |
| CD123 | 97% | Myeloid progenitors, plasmacytic dendritic cells, eosinophils, endothelial cells | 42 |
| Lewis Y (CD174) | 46% | Hematopoietic progenitor cells, intestinal epithelial cells | 47 |
| CD244 | 95% | NK cells, T cells, monocytes, basophils, eosinophils, spleen | 42 |
| TIM3 (CD366) | 85% | T cells, lung tissue | 42 |
| CLL-1 (CLEC12A, CD371) | 80% | Granulocytes, monocytes | 30,31 |
| Folate receptor B | 70% | Myeloid cells | 48 |
| NKG2DL | 70% | T-regulatory cells, endothelial cells | 49 |
NK, natural killer; T-ALL, T-cell acute lymphoblastic leukemia; T-LL, T-cell lymphoblastic lymphoma.
Leading immunotherapeutic targets in AML include CD33 and CD123. CD33 is expressed in a majority of AML cases and, given the pediatric experience with gemtuzumab ozogamicin, is a natural candidate for CAR T-cell therapy. Similarly, CD123 is an appealing target for CAR T-cell therapy, given the expanding landscape of CD123-targeting strategies. For both targets, there are several CAR T-cell trials either currently recruiting or in development that include pediatric participation (Table 4). Preliminary results from adult trials have been largely limited to case reports, but both targets appear to have shown benefit, at least in a subset of patients.27,28 There are numerous emerging targets, including Lewis Y antigen, NKG2DL, CD44v6, CLL-1, CD7, CD38, and FLT3, that may show similar benefit in a subset of patients (Table 3).29-31
Active chimeric antigen receptor T-cell clinical trials for acute myeloid leukemia
| Target | ClinicalTrials.gov identifier | Institution/sponsor | Age restriction | Status |
|---|---|---|---|---|
| CD7 | NCT04033302 | Shenzhen Geno-Immune Medical Institute | 6 mo-75 y | Recruiting |
| CD33 | NCT03971799 | Center for International Blood and Marrow Transplant Research | 1-30 y | Recruiting |
| CD44v6 | NCT04097301 | Horizon 2020/MolMed | 1-75 y | Recruiting |
| CD123 | NCT02159495NCT03766126NCT04109482NCT03190278NCT04318678 | City of Hope Medical CenterUniversity of PennsylvaniaMustang BioCellectisSt. Jude Children’s Research Hospital | >12 y>18 y>18 y18-64 y<21 y | RecruitingRecruitingRecruitingRecruitingRecruiting |
| NKG2D | NCT04167696NCT03018405 | Celyad OncologyCelyad Oncology | >18 y>18 y | RecruitingRecruiting |
| FLT3 | NCT03904069 | Amgen | >12 y | Not yet recruiting |
| CD123/CLL1 | NCT03631576 | Fujian Medical University | <70 y | Recruiting |
| CD123/CD33 | NCT04156256 | iCell Gene Therapeutics | None | Recruiting |
| CCL1/CD22/CD123 | NCT04010877 | Shenzhen Geno-Immune Medical Institute | 6 mo-75 y | Recruiting |
| Muc1/CLL1/CD22/CD38/CD56/CD123 | NCT03222674 | Shenzhen Geno-Immune Medical Institute | 2-75 y | Recruiting |
| CD33/CD28/CD56/CD123/CD117/CD133/CD34/Muc1 | NCT03473457 | Zhujiang Hospital | >6 mo | Recruiting |
| Target | ClinicalTrials.gov identifier | Institution/sponsor | Age restriction | Status |
|---|---|---|---|---|
| CD7 | NCT04033302 | Shenzhen Geno-Immune Medical Institute | 6 mo-75 y | Recruiting |
| CD33 | NCT03971799 | Center for International Blood and Marrow Transplant Research | 1-30 y | Recruiting |
| CD44v6 | NCT04097301 | Horizon 2020/MolMed | 1-75 y | Recruiting |
| CD123 | NCT02159495NCT03766126NCT04109482NCT03190278NCT04318678 | City of Hope Medical CenterUniversity of PennsylvaniaMustang BioCellectisSt. Jude Children’s Research Hospital | >12 y>18 y>18 y18-64 y<21 y | RecruitingRecruitingRecruitingRecruitingRecruiting |
| NKG2D | NCT04167696NCT03018405 | Celyad OncologyCelyad Oncology | >18 y>18 y | RecruitingRecruiting |
| FLT3 | NCT03904069 | Amgen | >12 y | Not yet recruiting |
| CD123/CLL1 | NCT03631576 | Fujian Medical University | <70 y | Recruiting |
| CD123/CD33 | NCT04156256 | iCell Gene Therapeutics | None | Recruiting |
| CCL1/CD22/CD123 | NCT04010877 | Shenzhen Geno-Immune Medical Institute | 6 mo-75 y | Recruiting |
| Muc1/CLL1/CD22/CD38/CD56/CD123 | NCT03222674 | Shenzhen Geno-Immune Medical Institute | 2-75 y | Recruiting |
| CD33/CD28/CD56/CD123/CD117/CD133/CD34/Muc1 | NCT03473457 | Zhujiang Hospital | >6 mo | Recruiting |
Summary of active and completed clinical trials according to www.clinicaltrials.gov as of 1 June 2020.
Because of the phenotypic heterogeneity of myeloblasts, none of the above antigens are likely to serve as universal targets, and targeting one alone may be insufficient to clear disease. It is reasonable to assume that antigen escape will emerge as a limitation of single-antigen–targeting CAR T cells in AML. Early results from CD123-specific CAR T cells in patients with AML demonstrate an efficacy signal without myeloablation but a lack of durable remission.27 Different genotypes of CD33 are widespread, and variants can cause a truncated CD33 protein to be expressed.32 There is a higher prevalence of these CD33 splice variants than of CD19, raising concerns for the frequency of antigen escape after CD33-directed CAR T-cell therapy. Given these concerns over the potential for antigen escape, most clinical trials in AML recommend consolidating a remission after CAR T cells with hematopoietic stem cell transplant (HSCT).
An alternative strategy to overcome this restricted antigen expression is through the use of dual targeting strategies. This is done by combining different targeting populations of CAR T cells, expressing more than one distinct CAR molecule on a single T cell, or engineering two different binding domains into a single CAR. Dual targeting allows multiple antigens to activate the cells synchronously. In theory, this could mitigate the risk of antigen escape and is currently being investigated in other hematologic malignancies.33 An added risk of a combinatorial targeting strategy is the possibility of increased on-target/off-tumor toxicity, and thorough investigations of individual targets is required before multiplexed targets.
A unique challenge to treating AML with CAR T cells is the growing evidence surrounding the immunosuppressive microenvironment resident to the bone marrow niche (Figures 1 and 2). Multiple groups have shown infiltration of regulatory T cells, myeloid-derived suppressor cells, and macrophages in the tumor microenvironment as well as coinhibitory checkpoint expression on effector T cells.34 This microenvironment appears to be as heterogeneous as the disease itself, with patients clustering into subgroups based on immune gene expression profiling.34 This immunosuppressive environment may further hinder our ability to provide effective cellular therapies by dampening CAR T-cell function. Alternatively, phenotyping this environment might serve as a biomarker to identify those patients who would benefit most from CAR T-cell therapy. Similarly, the immunosuppressive environment might represent a novel opportunity for enhancing the potency of CAR T cells via immune checkpoint blockade or other mechanisms.35
On-target/off-tumor toxicity
Unless a truly specific tumor antigen is targeted, there will always be a risk of on-target/off-tumor toxicity. A shared concern between T-cell and myeloid targeting strategies is the risk of prolonged immune suppression and life-threatening infections, with many groups exploring strategies to circumvent this challenge (Figures 1 and 2).
For T-cell and myeloid targeting, the ongoing persistence of CAR T cells may be detrimental. The B-cell aplasia seen with CD19 and CD22 targeting strategies is considered a permissive toxicity because it is readily managed with periodic immunoglobulin infusions. In contrast, prolonged T-cell or myeloid aplasia is a life-threatening immunodeficiency. The degree of myeloablation with different targets is yet to be defined and will require ongoing clinical investigations to understand the implications, including the time to recovery and/or ability to recover from myeloid cell aplasia with removal of the antigenic targeting. Early results of CD33/CLL-1 dual targeting therapy has demonstrated that, at least in some patients, complete myeloablation is noted and is used as a component of conditioning for HSCT.28 Conversely, early experience with CD123 CAR T cells has not resulted in treatment-related cytopenias.27 Therefore, strategic antigen selection, limitation of persistence, or rapid transition to HSCT may need to be employed to mitigate this risk.
One strategy is designing CAR T cells for transient in vivo persistence. This can be accomplished up front by using the more potent but less persistent CD28 costimulatory domain over the more persistent 41-BB costimulatory domain. Modulation of persistence can also be accomplished through the incorporation of suicide switches (eg, herpes simplex virus thymidine kinase, inducible caspase 9, coexpression of synthetic surface proteins targetable by monoclonal antibodies).36 Recovery from myeloid cell aplasia or T-cell depletion after CAR T-cell therapy can also be accomplished through a consolidative HSCT in which the CAR T cells are ablated.
An alternative to suicide switches or a consolidative HSCT is the ability to include “on-and-off” switches, which in theory can allow precise control of the CAR T-cell activity. For B-ALL, persistence has been demonstrated as a key attribute in the prevention of relapse.37 It is currently unclear how essential ongoing persistence of CAR T cells will be for non–B-cell hematologic malignancies, but one would hypothesize that it will be required for ongoing remission, thus limiting the applicability of CAR T-cell specifically engineered for limited in vivo persistence. By controlling the activity of the CAR, one could allow ongoing persistence while also managing the off-tumor toxicity. An example of this is the dimerizing agent–regulated immunoreceptor complex CAR T cells composed of separate antigen-targeting and signal transduction polypeptides that will only dimerize, and therefore function, in the presence of rapamycin.38 Finally, similar to the strategy of dual targeting antigens to enhance efficacy, there are strategies to combine an activating CAR that targets a tumor antigen with an inhibitory CAR that engages with an antigen on normal tissue, thereby reducing on-target/off-tumor toxicity.39 Until one of these strategies becomes consistently successful, CAR T-cell therapy for T-cell and myeloid disorders will likely serve as a bridge to an allogeneic HSCT to both reverse any ongoing on-target/off-tumor toxicity and increase the chance of cure.
Summary
Justifiable excitement exists surrounding successful integration of CAR T cells into the treatment of children and adolescents with high-risk non–B-cell hematologic malignancies. There are several unique barriers that exist which have limited implementation of this therapy, but novel strategies are currently in development and being investigated in clinical trials that seek to overcome these barriers. It is anticipated that several iterations will be required before we are able to parallel the success that has been seen in B-ALL.
Acknowledgments
A.J.L. was supported by the Seattle Children’s Hospital Cancer and Blood Disorders Center Research Pilot Program. R.G. was supported by Alex’s Lemonade Stand Foundation for Childhood Cancer, Stand Up To Cancer, St. Baldrick’s Foundation, and National Institutes of Health/National Center for Advancing Translational Sciences grant U01TR002487.
