
Abstract
Self-renewing hematopoietic stem cells and their progeny, lineage-specific downstream progenitors, maintain steady-state hematopoiesis in the bone marrow (BM). Accumulating evidence over the last few years indicates that not only primitive hematopoietic stem and progenitor cells (HSPCs), but also cells defining the microenvironment of the BM (BM niche), sense hematopoietic stress signals. They respond by directing and orchestrating hematopoiesis via not only cell-intrinsic but also cell-extrinsic mechanisms. Inflammation has many beneficial roles by activating the immune system in tissue repair and as a defense mechanism. However, chronic inflammation can have detrimental effects by stressing HSPCs, leading to cell (DNA) damage resulting in BM failure or even to leukemia. Emerging data have demonstrated that the BM microenvironment plays a significant role in the pathogenesis of hematopoietic malignancies, in particular, through disrupted inflammatory signaling, specifically in niche (microenvironmental) cells. Clonal selection in the context of microenvironmental alterations can occur in the context of toxic insults (eg, chemotherapy), not only aging but also inflammation. In this review, we summarize mechanisms that lead to an inflammatory BM microenvironment and discuss how this affects normal hematopoiesis. We pay particular attention to the process of aging, which is known to involve low-grade inflammation and is also associated with age-related clonal hematopoiesis and potentially malignant transformation.
Learning Objectives
Review the mechanisms leading to an inflammatory bone marrow (BM) microenvironment
Discuss the consequences of an inflammatory BM microenvironment on hematopoiesis and malignant transformation
Critically discuss the application of anti-inflammatory therapies
The BM microenvironment in steady state
A stem cell niche is defined as a local tissue microenvironment that ensures the maintenance and regulation of a stem cell or progenitor cells. The emerging picture of the bone marrow (BM) niche is complex and involves a multitude of cell types with important roles in osteogenesis, adipogenesis, and maintenance of the vasculature, mainly ensuring the maintenance of hematopoietic stem cell (HSC) function and the regulation of blood production. Evidence over the last years indicates that a dysfunction of the interaction between hematopoietic stem and progenitor cells (HSPCs) and distinct stem cell niche populations either in malignant hematopoietic diseases or also in inflammatory processes disrupts the tight regulation of hematopoiesis and favors a secretory phenotype of microenvironmental cells leading to impaired hematopoiesis-supporting capacity.1,2
Several stromal cell populations are involved in the regulation of hematopoiesis, and recent studies have dissected and defined the role of specific stromal cell populations on regulating distinct hematopoietic progenitor populations.3-5 BM mesenchymal stromal cells (BM-MSCs) are a rare population of cells in proximity to arterioles and sinusoid vessels (perivascular niche) or in proximity to the bone (endosteal niche).6 As an example, osteoblastic lineage cells (OBCs) express various factors important for HSC function (Winkler et al7 and Schepers et al8 ). Although MSCs are a heterogeneous population and still require better characterization by surface markers, perivascular mesenchymal cells that express CD146 in humans and Cxcl12–green fluorescent protein, Nestin-green fluorescent protein, full-length Leptin receptor, Prx-1–Cre, Osterix-Cre, and inducible Mx-1–Cre in mice generate osteoblastic cells and express factors that promote the maintenance of HSPCs.9 CXCL12 is essential in homing HSCs to the BM whereas KITL (also known as stem cell factor 1 [SCF-1]) supplies hematopoietic progenitors with an essential survival signal.10-12 Endothelial cells themselves are also a part of the perivascular HSC niche.13
A very specialized and well-studied hematopoietic niche is the erythroblastic island. Erythroblastic islands are very specialized niches supporting the proliferation and differentiation of erythroblasts.14 Erythroid islands are characterized by a central macrophage that extends cytoplasmic protrusions to a ring of adjacent erythroblasts. The interaction of cells within the island is crucial for early and late stages of erythroid maturation.
Furthermore, the nervous system is an important regulator of the niche, and already influences stem cell differentiation and maintenance early in development. Recent findings dissected and determined different modes of neural control, locally, in particular, by neural crest-derived MSCs, and intrinsically by hematopoietic cells that express neural receptors and neurotransmitters.15 Dysregulation between neural and hematopoietic systems can be induced by inflammation and subsequently contribute to disease, as discussed in the section “Inflammatory BM microenvironment and myeloid malignancies.” Other cell types that regulate HSC niches include osteoclasts, extracellular matrix, megakaryocytes, and calcium.9
Inflammation-induced activation of hematopoiesis
Inflammation is defined as a protective immune response as a consequence of pathophysiological processes initiated by, for example, infection and tissue injury/damage. The initial defense against infection is often initiated by innate immunity: pathogens are recognized by pattern recognition receptors (PRRs) expressed on hematopoietic and nonhematopoietic cells. In turn, innate immune cells then secrete an array of inflammatory cytokines and chemokines, tumor necrosis factor-α (TNF-α), as well as chemical mediators, for example, leukotriene B4, prostaglandin E2, and histamine upon ligation of PRRs by pathogen-derived molecules.16
A growing body of work has dissected the role of specific inflammatory signals, particularly cytokines and Toll-like receptor (TLR) ligands, in determining HSC function, maintenance, and blood output. Under normal conditions, HSCs are in a quiescent state. In response to inflammation, they rapidly start losing quiescence and transiently proliferate, also due to disturbed signals from their niche. In particular, the effect of interferons (IFNs) has been extensively studied in this context.17-19
IFN-γ, a prominent type II interferon, is produced during acute infections and leads to an activation and proliferation of otherwise quiescent HSCs to immunologically fight pathogens.20 Chronic exposure, however, functionally impairs HSCs in vitro and in vivo,21 in particular when secreted by stromal cells within the hematopoietic niche.22 IFN-γ mainly affects the self-renewal and repopulating capacity of hematopoietic progenitors by moderating their response to other cytokines.23 This effect is mainly mediated by STAT1 and suppressor of cytokine signaling 1 (SOCS1) and SOCS3 as effector molecules.21,24
Importantly, one should be mindful that IFN-γ is a pleiotropic mediator, upregulated in many different inflammatory settings. Its role is therefore closely intertwined with other proinflammatory signaling molecules. Hence, it is often difficult to distinguish the effect of these because they may be confounded by IFN responses. Furthermore, inflammatory signals induce lineage specification by different mechanisms in HSCs and impairs HSC self-renewal. Signaling through TLRs, TNF, and interleukin 1 (IL-1) negatively influences the long-term potential of HSCs through activation of apoptosis or induction of myeloid differentiation. Quiescence prevents HSC depletion during chronic inflammation. This indicates that multipotent progenitor (MPP) cells, responsible for daily blood maintenance, compensate the ongoing need, thereby minimizing the necessity for HSCs to proliferate.25
The inflammatory BM microenvironment
The interaction of HSCs and their progeny during stress hematopoiesis in inflammation with their microenvironment remains poorly defined. So far, mainly, the whole BM microenvironment is described as an inflammatory unit without specifically defining the different microenvironments/niches. Up to date, the best-studied microenvironment in inflammation is the erythroblastic island. Already described many decades ago as a functional unit, the erythroblastic island is moving into focus as being a central mediator of impaired erythropoiesis in various anemic disease.26 Inflammation, in particular with IL-6 as a central mediator, affects iron metabolism, thereby affecting hemoglobin synthesis and, hence, anemia.27 Interestingly, under inflammatory conditions, the central macrophage itself can become a source of inflammatory mediators. IFN-γ, secreted by the central macrophage, downregulates the Epo receptor on erythroid precursors, while at the same time upregulating PU.1. High levels of PU.1 cause a restriction of MPPs to granulocytic, monocytic, and lymphoid differentiation at the cost of megakaryocyte and erythroid differentiation, thus not only affecting erythroid differentiation but also myeloid cells.28 In concert, another prominent inflammatory cytokine, TNF-α, induces activation of caspases in erythroid precursor cells. The subsequent cleavage and depletion of GATA-1 as vital transcription factor for the erythroid lineage boosts the PU.1-mediated skewing.29,30 Additionally, upregulation of TNF family member proteins in nursing macrophages as well as maturing erythroblasts inhibits their differentiation even further and induces extrinsic apoptosis.31 As the interplay between macrophages and red blood cell progenitors is increasingly elucidated, more candidate proteins are identified as being involved in the pathophysiology of anemia.
Although spatial effects of inflammation in MSCs from different niches (endosteal and perivascular) have not been dissected specifically as compared with the interplay between the central macrophage and erythroid progenitor cells in the erythroid island, it has become increasingly clear that MSCs and endothelial cells (ECs) express various cytokine/chemokine receptors and PRRs. They sense an inflammatory milieu (locally or systemically) that is due to hematopoietic challenges. MSCs play a role in innate and acquired immunity directly through cell-cell contact or secretion of soluble factors.32,33 Thus, it is very likely that MSCs in both the endosteal and perivascular niche also acquire inflammatory programs and directly affect the maturation of neighboring HSPCs.
Additionally, inflammatory cytokines secreted from MSCs also affect more differentiated cells. IL-6, also secreted by MSCs, can induce an anti-inflammatory phenotype in macrophages. Granulocyte colony-stimulating factor (G-CSF), as a myeloid-supporting cytokine that is upregulated in infection, can enhance myelopoiesis through MSCs and suppress the production of CXCL12 from MSCs in the BM.34 Upon TLR stimulation, MSCs can induce the migration of monocytes from the BM by expressing the monocyte-recruiting chemokine, CCL2 also known as MCP1.35-37 In response to TLR4 activation, G-CSF is predominantly produced in ECs, and strongly stimulates emergency granulopoiesis with recruitment of neutrophils to the site of infection.38 These studies suggest that inflammation affects the cellular functions of MSCs and ECs, and influences hematopoiesis mainly through cytokine secretion or reaction to cytokines.39-41
Recent elegant single-cell studies resolved the effect of stress hematopoiesis on distinct, previously not described, niche cells in the BM. They provided single-cell resolution of distinct perivascular, vascular, and osteolineage compartments of the BM niche and match hematopoietic factors in stress hematopoiesis to their cellular origin. They particularly showed that the loss of the vascular-endothelial–expressed Notch ligand DLL4 skews HSPC differentiation toward myelopoiesis.42 Future studies using single-cell sequencing of microenvironmental cells and hematopoietic cells in inflammation and steady state will be instrumental in dissecting which niche cells contribute to inflammatory changes in the BM.
Aging and inflammation in the BM microenvironment
Importantly, a sustained inflammatory response might ultimately have detrimental effects on hematopoiesis, resulting in, for example, BM failure or even hematopoietic malignancies. The best-studied example is sustained IFN-α/γ activation during chronic infection, which impairs HSC function ultimately leading to BM failure.43 Along the same line, chronic TNF-α signaling was linked to development of myelodysplastic syndrome (MDS) and BM failure.44 Therefore, inflammatory responses must be terminated and the exact timing of this cessation is critical. Chronic inflammatory diseases can lead to a state of chronic inflammation. As chronic inflammatory diseases, including chronic infection, chronic kidney disease, autoimmune diseases, and chronic metabolic diseases (eg, atherosclerosis, type II diabetes), affect a large part of the elderly population and aging itself is also associated with chronic inflammation, it is important to understand the consequences of chronic inflammation on hematopoiesis and also the BM microenvironment. Inflammation in the natural aging process is reflected by anemia, immunosenescence, and thrombocytosis. In elderly patients, a systemic abundance of proinflammatory cytokines (IL-1, TNF, and IL-6) can be detected, which is called senescence-associated secretory phenotype (SASP).45 The SASP is most likely initiated by aging/senescent cells producing IL-1a in the microenvironment.46 Recent single-cell studies on niche cells in young and aged mice demonstrated activation of inflammatory response programs, changes in osteoblastic differentiation, and also metabolism with age, in particular through upregulation of IL1α/β.47 The pharmacological inhibition of IL-1 signaling resulted in recovery of the aged hematopoietic system from inflammatory (regenerative) stress. This study proposed that the inflammatory microenvironment in the old BM cavity drives remodeling of the niche and hematopoiesis. They proposed that blocking of proinflammatory cytokines is instrumental in reverting the aging of the BM niche and the impaired regenerative potential of old HSCs. Aging-associated (intrinsic) HSC alterations, including reduced self-renewal and myelopoiesis-skewed differentiation, strongly resemble the functional changes of HSCs exposed to chronic inflammation: increase of CD150high HSCs giving rise to predominantly myeloid lineage cells (myeloid skewing). The accumulation of myeloid-skewed HSCs in aging is mediated by activated signaling of TLR4, P-selectin, NF-κB, RANTES–mammalian target of rapamycin (mTOR), and transforming growth factor-β (TGF-β). Myeloid cells and adipocytes further produce numerous proinflammatory cytokines leading to a vicious cycle with accumulation of myeloid or myelodid-skewed cells in the BM.48
Thus, aging not only causes intrinsic changes in HSCs, but also induces significant changes in the BM microenvironment (leading to extrinsic mechanisms in HSC regulation). The aging of HSCs has been extensively studied. The mechanisms, however, of how aging affects the function and structure of the BM microenvironment are only recently being investigated. Recent evidence suggests that altered transcriptional landscapes, abnormal transduction of signaling cascades, epigenetic modifications, cytoskeletal polarity, cellular senescence, and clonal selection are key features of the aging process.
The relevance of niche interaction and signaling is highlighted by the fact that aged HSCs transplanted into a young BM environment exhibit less characteristics associated with senescent hematopoiesis.49 A frequent observation is the natural replacement of hematopoietic BM with adipose tissue in the elderly.50 This not only perturbs the mechanical stability of bone structure, but also immediately affects hematopoiesis because adipocytes could be identified as sources of regulatory cytokines. Through differentiation into mature adipocytes, multipotent MSCs lose their capacity to produce hematopoiesis supporting factors such as CXCL-12, IGF-1, and Kitl. The concentration of these is inversely correlated with the amount of BM fat.50 Moreover, adipocytes have been described to secrete mediators suppressing hematopoiesis and upholding a proinflammatory environment such as TNF-α.51 As the inflammatory pathways activated in aging also play a role in the pathogenesis of hematological disease, significant interest has emerged in studying the interaction between chronic inflammation, aging, and, ultimately, hematological malignancy (Table 1). In particular, the SASP-associated cytokines TNF, IFN, and IL-6 all play significant roles in the pathogenesis of myeloproliferative neoplasms (MPNs),52 MDSs,53 and acute myeloid leukemia (AML).54
Inflammatory factors in the microenvironment with implication in the development of hematopoietic malignancies
| Effector molecules/cellular source | Target cells | Cellular changes | Functional effect | Reference | Year |
|---|---|---|---|---|---|
| Arg1 | |||||
| MDSCs, MSCs | Arg depletion, TCR ↓ | TC anergy | 90 | 2009 | |
| IDO1/2 | CD8+ TCs, NKCs, NK-TCs | Trp depletion, mTOR activation | Proliferation arrest | 91 | 2012 |
| Production of kynurenine derivatives, AhR activation | Treg differentiation | 92 | 2010 | ||
| iNOS | TCs | NO production | Treg differentiation | 93 | 2006 |
| IL-10 | |||||
| MDSCs, MSCs | Macrophages, DCs | Inhibition of APCs | 94 | 2015 | |
| TGF-β | |||||
| MDSCs, megakaryocytes | CD4+ TCs | Foxp3 ↑ | Treg differentiation | 95 | 2010 |
| Schwann cells | IL-12Rβ2 ↓ | Inhibition of Th1 differentiation | |||
| GATA-3 ↓ STAT6↓ | Inhibition of Th2 differentiation | ||||
| S100A8 | |||||
| Neutrophils, monocytes | MDSCs | CD33 activation | MDSC expansion | 96 | 2008 |
| Macrophages, MDSCs | Sustained inflammatory response | 75 | 2019 | ||
| HSPCs | TLR4 activation, p53 activation, NFκB activation, ROS ↑, DNA damage response | HSPC loss and leukemic evolution | 68 | 2016 | |
| Erythroid precursors | TLR4 activation, p53 activation | Erythroid differentiation defect | 66 | 2016 | |
| MSCs | Loss of hematopoiesis support | 70 | 2019 | ||
| IFN-γ | |||||
| Th1 cells, macrophages | HSCs | STAT1 activation, SOCS1/SOCS3 activation | Responsiveness to growth factors ↓ | 23 | 2014 |
| CD8+ T cells | Fas↑, caspase activity ↑ | Self-renewal, repopulation capacity ↓ | |||
| Erythroid precursors | EpoR↓ PU.1↑ | Myeloid priming of MPPs, erythroid differentiation ↓ | 28 | 2007 | |
| MSCs | IL-6 induction | 97 | 2014 | ||
| TNF-α | |||||
| Adipocytes, macrophages | MPPs | Caspase-1 induction → GATA-1 cleavage | 30 | 2005 | |
| IL-6 | |||||
| MSCs, macrophages | Hepatocytes | Hepcidin induction → iron depletion | Hb biosynthesis ↓ | 27 | 2005 |
| EC | G-CSF ↑ | 40 | 1986 | ||
| G-CSF | |||||
| EC | MSCs | Stimulation of myelopoiesis | 39 | 2014 | |
| Inhibition of lymphopoiesis | |||||
| CXCL12 ↓ mobilization | 34 | 2012 | |||
| IL-1α/β | |||||
| DCs, monocytes | Sympathetic nerve fibers | Sympathetic neuropathy | 81 | 2014 | |
| Macrophages | 80 | 2014 | |||
| Senescent cells | SASP induction | 46 | 2009 | ||
| 45 | 2010 | ||||
| HSCs | TLR4 signaling, p38-MAPK activation | 98 | 2017 |
| Effector molecules/cellular source | Target cells | Cellular changes | Functional effect | Reference | Year |
|---|---|---|---|---|---|
| Arg1 | |||||
| MDSCs, MSCs | Arg depletion, TCR ↓ | TC anergy | 90 | 2009 | |
| IDO1/2 | CD8+ TCs, NKCs, NK-TCs | Trp depletion, mTOR activation | Proliferation arrest | 91 | 2012 |
| Production of kynurenine derivatives, AhR activation | Treg differentiation | 92 | 2010 | ||
| iNOS | TCs | NO production | Treg differentiation | 93 | 2006 |
| IL-10 | |||||
| MDSCs, MSCs | Macrophages, DCs | Inhibition of APCs | 94 | 2015 | |
| TGF-β | |||||
| MDSCs, megakaryocytes | CD4+ TCs | Foxp3 ↑ | Treg differentiation | 95 | 2010 |
| Schwann cells | IL-12Rβ2 ↓ | Inhibition of Th1 differentiation | |||
| GATA-3 ↓ STAT6↓ | Inhibition of Th2 differentiation | ||||
| S100A8 | |||||
| Neutrophils, monocytes | MDSCs | CD33 activation | MDSC expansion | 96 | 2008 |
| Macrophages, MDSCs | Sustained inflammatory response | 75 | 2019 | ||
| HSPCs | TLR4 activation, p53 activation, NFκB activation, ROS ↑, DNA damage response | HSPC loss and leukemic evolution | 68 | 2016 | |
| Erythroid precursors | TLR4 activation, p53 activation | Erythroid differentiation defect | 66 | 2016 | |
| MSCs | Loss of hematopoiesis support | 70 | 2019 | ||
| IFN-γ | |||||
| Th1 cells, macrophages | HSCs | STAT1 activation, SOCS1/SOCS3 activation | Responsiveness to growth factors ↓ | 23 | 2014 |
| CD8+ T cells | Fas↑, caspase activity ↑ | Self-renewal, repopulation capacity ↓ | |||
| Erythroid precursors | EpoR↓ PU.1↑ | Myeloid priming of MPPs, erythroid differentiation ↓ | 28 | 2007 | |
| MSCs | IL-6 induction | 97 | 2014 | ||
| TNF-α | |||||
| Adipocytes, macrophages | MPPs | Caspase-1 induction → GATA-1 cleavage | 30 | 2005 | |
| IL-6 | |||||
| MSCs, macrophages | Hepatocytes | Hepcidin induction → iron depletion | Hb biosynthesis ↓ | 27 | 2005 |
| EC | G-CSF ↑ | 40 | 1986 | ||
| G-CSF | |||||
| EC | MSCs | Stimulation of myelopoiesis | 39 | 2014 | |
| Inhibition of lymphopoiesis | |||||
| CXCL12 ↓ mobilization | 34 | 2012 | |||
| IL-1α/β | |||||
| DCs, monocytes | Sympathetic nerve fibers | Sympathetic neuropathy | 81 | 2014 | |
| Macrophages | 80 | 2014 | |||
| Senescent cells | SASP induction | 46 | 2009 | ||
| 45 | 2010 | ||||
| HSCs | TLR4 signaling, p38-MAPK activation | 98 | 2017 |
AhR, aryl hydrocarbon receptor; APC, antigen-presenting cell; Arg1, arginase 1; DC, dendritic cell; Hb, hemoglobin; IDO, indoleamine 2, 3-dioxygenase; iNOS, inducible nitric oxide synthase; MDSC, myeloid-derived suppressor cell; NKC, natural killer cell; NKT, natural killer T cell; NO, nitric oxide; ROS, reactive oxygen species; TC, T cell; TCR, T-cell receptor; Th, T helper; Treg, regulatory T cell; Trp, tryptophan.
Aplastic anemia: BM failure caused by inflammation
The prototypical example of a hematological disease caused by BM inflammation is aplastic anemia (AA). Although the pathogenesis is still not entirely understood, type I IFNs have been identified to be involved in the development of AA.55 IFN-γ can directly affect HSCs: STAT1 signaling leads to cycling and differentiation, ultimately resulting in stem cell exhaustion, whereas upregulation of SOCS1 blocks the self-renewing capacities.56 Indirect effects of IFN-γ on macrophages and MSCs have been implied to contribute to high levels of accessory inflammatory cytokines like IL-6, IL-1β, and TNF-α.57 It is important to note that the inflammatory processes in AA predominantly affect HSCs and not the stromal cells. Stromal cells isolated from aplastic BM have the capacity to form a functional hematopoietic microenvironment in vivo.58 Moreover, allogenic stem cell transplantation with a reduced-intensity conditioning regimen, which is thought to preserve the host stroma, is a successful therapy for reinstating hematopoiesis in eligible patients.59
Another major effector of damage is a type 1 T-helper (Th1) cell–skewed response of the adaptive immune system, which was also identified as the major source of IFN-γ, accompanied by a reduction in regulatory T cells (Tregs) in the marrow.60 In the majority of AA patients, glycosylphosphatidylinositol-specific T cells can be detected.61,62 A CD1-restricted T-cell response can lead to an elimination of hematopoietic cells, while at the same time creating a selective advantage for clones with deficiency for phosphatidylinositol glycan biosynthesis class A protein, reported in patients with paroxysmal nocturnal hemoglobinuria (PNH).63 Thus, the coincidence of PNH with AA provides an escape mechanism wherein the PNH clone can restore hematopoiesis by evading immune-mediated eradication. Other mutations commonly found in clonal hematopoiesis can simply be selected due to the overall BM hypocellularity.64 Thus, hypocellular MDS can be understood as an exhausted form of AA in which selective clones have been established.65
Inflammatory BM microenvironment and myeloid malignancies
It has become evident over the last several years that hyperactive innate immune signaling (eg, via the myeloid differentiation primary response 88/IL-1 receptor–associated kinase pathway and TNF receptor–associated factor 6, representing downstream signaling of innate immune inputs including TLRs) has emerged as a key driver in myeloid malignancies and also leukemic progression.
Whether and how chronic inflammation and thus aging induce leukemic transformation and attrition of (nonmalignant) HSCs remains an open question. One hint might be that HSC attrition or leukemic transformation has been observed when inflammation is accompanied by cell-intrinsic DNA repair defects in HSCs or further fueled by dysregulated mechanisms in the BM microenvironment, in particular inflammation, particularly in niche cells. The aberrant production of inflammatory signals in niche cells such as IFN, IL-1, and particularly in alarmins S100A8 and S100A9 (damage associated molecular pattern that signals via TLR4) was also shown to significantly contribute to myeloid disease.8,52,66-68 These alarmins exert specific effects on HSPCs depending on different niches of the BM. Increased S100A8/S100A9 levels in the BM are linked to anemia and an increase in monocytes. Furthermore, the aging BM microenvironment is composed of increasing amounts of damage-associated molecular patterns.69 In our previous work, we demonstrated that Rps14 haploinsufficiency in a murine model of del(5q) MDS leads to increased expression of S100A8 and S100A9 in mutant erythroblasts, monocytes, and macrophages, the main regulatory cells of the erythroid niche.70 We demonstrated specifically that S100A8 is responsible for the erythroid-differentiation defect as addition of recombinant S100A8 was sufficient to induce a differentiation defect in wild-type erythroid cells, and genetic inactivation of S100A8 expression rescued the erythroid-differentiation defect of Rps14-haploinsufficient HSCs. Interestingly, we also observed increased expression of S100A8/S100A9 in the mesenchymal niche in del(5q) MDS.70 Raaijmakers and colleagues confirmed these results and additionally showed increased levels of S100A8 in the mesenchymal niche in MDS and Shwachman-Bodian-Diamond syndrome, which affects HSCs through excessive reactive oxygen species (ROS) production and genotoxic stress.68,71 These studies demonstrate that inflammation in distinct (spatial) niches of the BM have distinct effects on hematopoiesis.
Another prominent aspect in MDSs is the presence of heterogeneous myeloid-derived suppressor cells (MDSCs). Under perpetuated inflammatory conditions, early granulocytic or monocytic precursors (not derived from the dysplastic clone) differentiate into granulocytic or polymorphonuclear MDSCs and monocytic MDSCs, respectively.72-74 On the one hand, these cells produce a plethora of anti-inflammatory mediators. On the other hand, MDSCs can contribute to the prevalent inflammation by secreting proinflammatory factors like S100A8/A9. Secreted S100 proteins cause autocrine activation and expansion of MDSCs via binding of CD33/Siglec-3.75 MDSCs are sufficient to create an MDS phenotype in murine experiments, and targeting them or their expansion leads to reconstitution of normal hematopoiesis.76,77
In summary, we know that the BM microenvironment is abnormal in myeloid malignancies, and exhibits great similarities to an inflammatory state. Whether this is a direct consequence of changes in hematopoietic cells or is instead independent of the disease (and caused by aging, chronic inflammation) is one of the main open questions in the field as stated in the introductory section. As increased levels of S100A8 in myeloid disease were noted not only in macrophages/monocytes (cells of the innate immune response) but also in mesenchymal cells, we wondered whether there was a direct interaction between those cells. Using an MDS mouse model and patient samples, we demonstrated that increased expression of S100A8 in MDS-derived macrophages induces S100A8 expression in neighboring MSCs. We further directly linked the increased expression of S100A8 in stromal cells to loss of their hematopoiesis-supporting capacity.70 These data indicate that intrinsic defects of MDS hematopoietic cells directly alter the surrounding microenvironment, which in turn negatively affects hematopoiesis as an extrinsic mechanism. We thus hypothesize that genetic alterations in hematopoietic cells lead to an altered inflammatory response in macrophages as part of the mutated clone and that these changes actively contribute to disease progression and ineffective hematopoiesis. The open question of whether niche abnormalities in myeloid malignancies are the hen or the egg, however, remains and is most likely context dependent. Molecular alterations in niche cells by themselves have been shown to induce myeloid malignancies.78 Thus, the conclusion can only be that the microenvironment is massively “reprogrammed” in myeloid malignancies and is inflammatory, leading to loss of function of the microenvironment.
A “reprogrammed” inflammatory microenvironment is a common theme in myeloid malignancies. Studies focusing on different myeloid diseases and also different niche cells were able to mechanistically elucidate how malignant, mutated hematopoietic cells and also proinflammatory cytokines activate distinct subsets of stromal cells in the BM, increase their secretory activity, and influence their reduced hematopoiesis-supporting capacity.1,2,6,79 In murine models of MPN80 and AML,81 the hematological malignancy created neuropathic changes in the BM niche, which affected the activity of MSCs and altered the function of the HSC niche, in particular leading to downregulated expression of many HSC-retention factors, including CXCL12 and SCF, indicating loss of hematopoiesis support. Schepers et al demonstrated that OBCs expand in the presence of malignant hematopoietic cells resulting in extensive remodeling of the BM microenvironment.8 They demonstrated the accumulation of inflammatory myelofibrotic cells in this context and downregulation of factors regulating the niche function and HSC retention, including the chemokine CXCL12, n-cadherin, SCF, angiopoietin-1, and TGF-1 and -2. These data indicate that malignant hematopoietic cells diminish the hematopoietic-supportive capacity of BM stromal and niche cells indirectly through changes in the physical and cytokine environment. Also, our own data on the role of stromal (Gli1+) MSCs in MPN showed altered expression of many HSC-regulatory genes and cytokines, including a broad downregulation of HSC-retention factors, in particular CXCL12, in myelofibrosis, indicating transcriptional reprogramming from hematopoiesis support to inflammatory (profibrotic) programs. In summary, both senescence/aging and other dysfunctions of the BM niche caused by chronic disease, infection, or aging may lead to overproduction of inflammatory signals that affect HSPCs and alter the fitness of their regulation microenvironment, which might lead to a microenvironment supporting transformed clones and not normal hematopoiesis, leading to a vicious cycle that further promotes the expansion of the malignant clone but not normal hematopoiesis.
Macrophages and monocytes, as cells of the innate immune response, are increased in chronic inflammatory states in aging and in myeloid malignancies and might be an important contributor to the remodeling of the BM microenvironment in inflammation and hematopoietic disease (anemia, BM failure, and also leukemia).70 Previous studies demonstrated that macrophages regulate HSPCs indirectly through interaction with MSCs.82 These findings are supported by recent findings demonstrating a causal link between mutations in HSPCs in the context of clonal hematopoiesis with indeterminate origin and altered transcriptional output of macrophages, which affect the vascular wall and contributes to atherosclerosis.83,84 Furthermore, inflammation driving expansion of resistant clones is beginning to be documented in the case of TET2 mutations. Tet2-knockout murine and TET2-mutant human HSPCs have a clonal advantage caused by resistance to apoptosis in an in vitro environment that contains the proinflammatory cytokine TNFα.85 TET2-deficient macrophages are hyperinflammatory, and this may exacerbate processes by propagating an inflammatory environment. Furthermore, increased IL-6 production or activation of TLR2 is critical for the development of the myeloproliferative phenotype of TET2-deficient hematopoietic cells. Future research will focus on the functional and molecular dissection of macrophages in myeloid disease, using tools such as single-cell RNA sequencing to determine subsets of macrophages that are mainly responsible for the inflammatory phenotype and whether the changes in macrophages/monocytes in inflammation, aging, and malignancies are the same or distinct in the different processes. Together, the data point to necessary contributions of the aberrant (inflammatory) microenvironment to drive the induction, selection, and eventual expansion of mutant clones that are poised for subsequent malignant transformation.
Importantly, malignant cells use elaborate ways to shield themselves from immune response. The mechanisms of immune evasion are as heterogeneous as AML itself and dependent on the differentiation stage of AML blasts. There is a wide overlap with evasion strategies observed in solid tumors. AML can lead to the induction of MDSCs and Tregs, which dampen adaptive immune response in close proximity to leukemic cells.32 Blasts and malignant driver cells in MPNs have also been described to upregulate B7-family proteins or programmed death-ligand 1 (PD-L1), inhibiting T cells through interaction with CTLA-4 and PD-1, respectively.86-88 Discussing the various mechanisms of immunoevasion in malignant cells would be out of the scope of this review. However, it is important to note that malignant cells have a higher capacity to resist inflammatory and immunological responses compared with physiological hematopoietic cells.
It is important to note that although clonal hematopoiesis is observed with increasing frequency with age, not all aging individuals develop hematologic malignancies or BM failure. The reasons for this are not clear, but likely involve interactions between cell-intrinsic and microenvironmental mechanisms as well as the genetic/epigenetic factors that regulate them. Given the emerging evidence for the microenvironment as a regulator of many aspects of HSC function as outlined herein, it would be interesting to determine the extent to which the microenvironment plays a modifying role in hematopoietic aging. Such studies might include a closer dissection of hematopoiesis and the BM microenvironment in patients with chronic inflammatory illnesses or after BM transplantation using donors and/or recipients of varying ages.
In summary, inflammation in the BM microenvironment seems to be a common stress response to various stimuli. It becomes increasingly clear that inflammatory signaling in the microenvironment is deeply intertwined with the function of HSPCs and affecting fate decisions. Inflammation acts as a double-edged sword, promoting normal HSC development and function in acute stress situations while promoting HSC deregulation and functional decline and even malignancy if sustained (Figure 1).
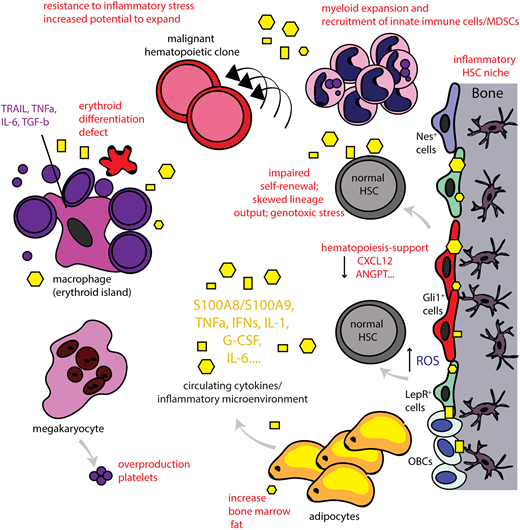
The inflammatory BM microenvironment. Disease-related chronic inflammation and/or physiological aging leads to the presence of continuous production of proinflammatory signals (systemically and locally in the BM). In turn, this leads to an inflammatory BM microenvironment as MSCs of the BM, for example, nestin+ (Nes+), Gli1+, leptin-receptor (LepR+) cells, acquire an inflammatory, secretory phenotype as well and also release proinflammatory signals that affect both the HSC niche and also the erythroblastic islands. This leads to significant alterations in HSC function and output. Myeloid cells expand and innate immune cells/MDSCs are recruited. High numbers of platelets are released by megakaryocytes whereas lymphoid cells decrease. The erythroid differentiation is significantly impaired leading to anemia (anemia of chronic inflammation or anemia in the elderly). The central macrophage of the erythroblastic island can also acquire an inflammatory phenotype and further contributes to inflammation in the BM microenvironment. Adipocytes are increased in the aged BM and can also release inflammatory signals. Chronic inflammation leads to loss of support of normal hematopoiesis and can lead to decreased self-renewal of HSCs and skewed lineage output. The continuous exposure to this stress situation leads to dysfunction of the BM microenvironment and increased reactive oxygen levels (ROS), inducing genotoxic stress for HSCs, potentially promoting genomic instability. Normal hematopoiesis may thus also be impaired in a manner such that preexisting HSC clones carrying potentially leukemic mutations have increased potential to expand and evolve. Thus, chronic inflammation in the BM microenvironment may function as an initiator or even driver of hematological malignancies. Yellow geometrical forms highlight inflammatory cytokines that are further specified in Table 1.
The inflammatory BM microenvironment. Disease-related chronic inflammation and/or physiological aging leads to the presence of continuous production of proinflammatory signals (systemically and locally in the BM). In turn, this leads to an inflammatory BM microenvironment as MSCs of the BM, for example, nestin+ (Nes+), Gli1+, leptin-receptor (LepR+) cells, acquire an inflammatory, secretory phenotype as well and also release proinflammatory signals that affect both the HSC niche and also the erythroblastic islands. This leads to significant alterations in HSC function and output. Myeloid cells expand and innate immune cells/MDSCs are recruited. High numbers of platelets are released by megakaryocytes whereas lymphoid cells decrease. The erythroid differentiation is significantly impaired leading to anemia (anemia of chronic inflammation or anemia in the elderly). The central macrophage of the erythroblastic island can also acquire an inflammatory phenotype and further contributes to inflammation in the BM microenvironment. Adipocytes are increased in the aged BM and can also release inflammatory signals. Chronic inflammation leads to loss of support of normal hematopoiesis and can lead to decreased self-renewal of HSCs and skewed lineage output. The continuous exposure to this stress situation leads to dysfunction of the BM microenvironment and increased reactive oxygen levels (ROS), inducing genotoxic stress for HSCs, potentially promoting genomic instability. Normal hematopoiesis may thus also be impaired in a manner such that preexisting HSC clones carrying potentially leukemic mutations have increased potential to expand and evolve. Thus, chronic inflammation in the BM microenvironment may function as an initiator or even driver of hematological malignancies. Yellow geometrical forms highlight inflammatory cytokines that are further specified in Table 1.
Inflammatory BM microenvironment: Do I treat? How to treat? When to treat?
A critical next step is the translation of these mostly basic research findings of inflammation in the BM microenvironment into clinical practice. As the overall contribution to pathogenesis and progression varies between diseases and also depends on the specific hematopoietic niches involved, treatment will differ between the discussed disease entities. For AA, which is predominantly mediated by T effector cells and their secreted cytokines, immunosuppressive therapy with ciclosporin and antithymocyte globulin to deplete the T-cell response is an established strategy with good success rates.89
For other diseases, defined by a less specific inflammatory BM microenvironment, intervention does not seem to be as straightforward. Although affecting hematopoiesis, aging-induced inflammation can be considered a physiological process and it is not clear whether anti-inflammatory treatment without any signs of malignancy of dysplasia in the BM were beneficial. Cutoff values for such treatments could be hemoglobin levels or erythrocyte counts as the erythroid system in particular is very sensitive to inflammation and is in a majority of cases affected in MDSs. Effective use of cytokine blockade and/or anti-inflammatory and antisenescence therapies to restore “normal (young)” hematopoiesis, improve erythropoiesis, and/or uniquely target malignant cells requires a deeper biological understanding of how inflammatory signals lead to blood system dysfunction and participate in the pathogenesis of hematological malignancy. However, the data summarized herein point to necessary contributions of the aberrant (inflammatory) microenvironment to drive the induction, selection, and eventual expansion of mutant clones that are poised for subsequent malignant transformation. The data strongly argue that myeloid malignancies needs to be considered as a systemic disease including both hematopoietic cells and their different microenvironment characterized by distinct functions. Cross talk between HSPCs and their microenvironment contributes to disease initiation and evolution. This has important consequences for our thinking about prognostication and treatment of this disease. Niche parameters should be considered that complement HSPC-intrinsic characteristics in future models of prognostication and target environmental contributions. Targeting could, to name just a few examples, involve inhibition of inflammation (in the niche and the hematopoietic system) through inhibition of TLR4 signaling, ROS, or the NLRP3 inflammasome, or even targeting of S100A8/S100A9. In sum, the questions (1) “Do I treat?,” (2) “How to treat?,” and (3) “When to treat?” cannot yet be answered sufficiently and probably will vary depending on the underlying disease and the contribution of inflammation to pathogenesis. However, anti-inflammatory strategies are finding their way into clinical practice.53,66-68,75 The results of randomized clinical trials will illuminate their potential benefits in the future.
Correspondence
Rebekka K. Schneider, Erasmus MC, Faculty Bldg 1330e, Wytemaweg 80, 3015 CN, Rotterdam, The Netherlands; e-mail: r.k.schneider@erasmusmc.nl.
