
Abstract
Patients with cancer have an increased risk of thromboembolism, which is the second leading cause of death in these patients. Several mechanisms of the prothrombotic state in these patients have been proposed. Among them are a platelet activation receptor, C-type lectin-like receptor 2 (CLEC-2), and its endogenous ligand podoplanin, which are the focus of this review. CLEC-2 is almost specifically expressed in platelets/megakaryocytes in humans. A membrane protein, podoplanin is expressed in certain types of cancer cells, including squamous cell carcinoma, brain tumor, and osteosarcoma, in addition to several normal tissues, including kidney podocytes and lymphatic endothelial cells but not vascular endothelial cells. In the bloodstream, podoplanin induces platelet activation by binding to CLEC-2 and facilitates hematogenous cancer metastasis and cancer-associated thrombosis. In an experimental lung metastasis model, the pharmacological depletion of CLEC-2 from platelets in mice resulted in a marked reduction of lung metastasis of podoplanin-expressing B16F10 cells. Control mice with B16F10 orthotopically inoculated in the back skin showed massive thrombus formation in the lungs, but the cancer-associated thrombus formation in CLEC-2–depleted mice was significantly inhibited, suggesting that CLEC-2–podoplanin interaction stimulates cancer-associated thrombosis. Thromboinflammation induced ectopic podoplanin expression in vascular endothelial cells or macrophages, which may also contribute to cancer-associated thrombosis. CLEC-2 depletion in cancer-bearing mice resulted in not only reduced cancer-associated thrombosis but also reduced levels of plasma inflammatory cytokines, anemia, and sarcopenia, suggesting that cancer-associated thrombosis may cause thromboinflammation and cancer cachexia. Blocking CLEC-2–podoplanin interaction may be a novel therapeutic strategy in patients with podoplanin-expressing cancer.
Learning Objectives
Explain general mechanisms of cancer-associated thrombosis
Explain how the platelet activation receptor CLEC-2 and podoplanin facilitate cancer-associated thrombosis
General introduction for cancer-associated thrombosis
Patients with cancer have an increased risk of thromboembolism, which is associated with substantial morbidity and mortality. The incidence of venous thromboembolism (VTE) has been reported to be 10% to 20% of patients with cancer; although less frequent, arterial thromboembolism (ATE) is also observed in 1% to 4.7% of patients with cancer.1,2 Although this relationship between cancer and thromboembolism has been recognized as Trousseau syndrome for 150 years, the mechanisms of frequent thromboembolism in cancer have not been fully elucidated. Several direct and indirect mechanisms, which differ depending on the types of cancer cells involved, have been proposed. Tumor cells directly activate the coagulation cascade. Pancreatic cancer cells upregulate the tissue factor (TF) and release TF-positive macrovesicles, which results in the direct initiation of the coagulation cascade.1 TF activates the extrinsic pathway of the coagulation cascade, resulting in thrombin generation and clot formation. Thrombin also strongly activates platelets. Plasminogen activator inhibitor (PAI)-1 inhibits fibrinolysis; therefore, increased levels of PAI-1 are associated with thrombosis. Several types of cancer cells produce PAI-1, which results in cancer-associated thrombosis. Furthermore, increased plasma levels of PAI-1 have been reported in patients with pancreatic cancer or brain tumor.1
Cancer cells also directly activate platelets by secreting platelet agonists adenosine 5′-diphosphate and thrombin.3 Mucinous adenocarcinomas secrete abnormally glycosylated mucins and mucin fragments into the bloodstream. These mucins have binding sites for selectins and interact with leukocyte L-selectin and platelet P-selectin, which results in the activation and aggregation of platelets.4,5 As discussed later, cancer cells themselves activate platelets by the interaction between a membrane protein on the surface of cancer cells and its specific receptor on platelets.
Cancer cells facilitate thrombus formation by indirectly acting on endothelial cells or leukocytes. They release proinflammatory cytokines, including interleukin (IL)-1β and tumor necrosis factor (TNF)-α, which promote prothrombotic change in endothelial cells.3 Neutrophil activation induces the release of decondensed chromatin fibers and granule proteins, which are called “neutrophil extracellular traps” (NETs). NETs not only kill bacteria but also activate platelets and coagulation factors. It has recently been reported that cancer cells facilitate NET formation and cancer-associated thrombosis and that a NET marker, citrullinated histone H3, predicts the risk of VTE and mortality in patients with cancer.6,7 Cancer cells induce granulocytosis and thrombocytosis by secreting granulocyte colony-stimulating factor and IL-6. (IL-6 stimulates thrombopoietin production in the liver.) These mechanisms are summarized in Table 1.
Direct and indirect mechanisms involved in cancer-associated thrombosis
| Target | Cancer cell–derived factor to induce prothrombotic status | Action | |
|---|---|---|---|
| Coagulation cascade | Increased TF expression | Thrombin and fibrin generation Platelet activation by thrombin | |
| PAI-1 | Inhibited fibrinolysis | ||
| Platelets | ADP release | Platelet aggregation via P2Y1 and P2Y12 and adhesion to cancer cells | |
| Podoplanin | Platelet aggregation via CLEC-2 and adhesion to cancer cells | ||
| Other | Neutrophils | Any factors derived from cancer | NET generation and subsequent activation of platelets and FXII by NETs |
| Platelets and neutrophils | IL-6 (stimulate TPO generation in the liver), G-CSF secretion | Granulocytosis and thrombocytosis | |
| Mucins | Aggregation via P-selectin in platelets and L-selectin in neutrophils | ||
| Endothelial cells | Inflammatory cytokines | Prothrombotic change | |
| Target | Cancer cell–derived factor to induce prothrombotic status | Action | |
|---|---|---|---|
| Coagulation cascade | Increased TF expression | Thrombin and fibrin generation Platelet activation by thrombin | |
| PAI-1 | Inhibited fibrinolysis | ||
| Platelets | ADP release | Platelet aggregation via P2Y1 and P2Y12 and adhesion to cancer cells | |
| Podoplanin | Platelet aggregation via CLEC-2 and adhesion to cancer cells | ||
| Other | Neutrophils | Any factors derived from cancer | NET generation and subsequent activation of platelets and FXII by NETs |
| Platelets and neutrophils | IL-6 (stimulate TPO generation in the liver), G-CSF secretion | Granulocytosis and thrombocytosis | |
| Mucins | Aggregation via P-selectin in platelets and L-selectin in neutrophils | ||
| Endothelial cells | Inflammatory cytokines | Prothrombotic change | |
ADP, adenosine 5′-diphosphate; CLEC-2, C-type lectin-like receptor 2; FXII, factor XII; G-CSF, granulocyte colony-stimulating factor; IL-6, interleukin 6; NET, neutrophil extracellular trap; PAI-1, plasminogen activator inhibitor 1; TF, tissue factor; TPO, thrombopoietin.
Clinical risk factors that contribute to the high incidence of cancer-associated thrombosis should be noted. Frequent hospitalization, immobility after surgery, and extrinsic compression of blood vessels by tumors cause venous stenosis. Major surgery and chemotherapy, including hormone therapy, lead to hypercoagulability. Central venous catheters and direct tumor invasion cause endothelial cell injury.2 Thus, patients with cancer are prone to develop a prothrombotic state, owing to both cancer pathology–related factors and cancer therapy–related factors.
It has been reported that thromboembolism is a leading cause of death in patients with cancer receiving outpatient chemotherapy.8 In addition, platelet activation and hypercoagulation induced by cancer cells actively contribute to tumor development and dissemination.9,10 For these reasons, thromboprophylaxis in patients with cancer is of potential clinical value. For many years, long-term therapy with low-molecular-weight heparin was the standard of care for the management of cancer-associated VTE.11 However, the Hokusai Cancer Venous Thromboembolism trial showed that for treatment or secondary prevention of cancer-associated VTE, a direct oral anticoagulant, edoxaban, was noninferior to subcutaneous dalteparin (a low-molecular-weight heparin) with respect to the composite outcome of recurrent VTE or major bleeding.12 The rate of recurrent VTE was lower, but the rate of major bleeding was higher, with edoxaban than with dalteparin.12 The SELECT-D (Anticoagulation Therapy in Selected Cancer Patients at Risk of Recurrence of Venous Thromboembolism) trial reported similar results.13 For primary prevention of VTE in patients with cancer, the AVERT (Apixaban for the Prevention of Venous Thromboembolism in Cancer Patients) trial reported that apixaban (a direct oral anticoagulant) therapy resulted in a significantly lower rate of VTE than did placebo among intermediate- to high-risk ambulatory patients with cancer who were starting chemotherapy. The rate of major bleeding episodes was higher with apixaban than with placebo.14 Another clinical trial for primary prevention of VTE in ambulatory patients with cancer receiving chemotherapy, the CASSINI (Efficacy and Safety of Rivaroxaban Prophylaxis Compared With Placebo in Ambulatory Cancer Patients Initiating Systemic Cancer Therapy and at High Risk for Venous Thromboembolism) trial, reported that rivaroxaban also reduced the incidence of VTE, although there was no statistical significance.15 It is suggested that these anticoagulant therapies inhibit thrombin, which is generated through TF-bearing cancer cells and/or various causes related to cancer therapy, and subsequently generate fibrin and activate platelets, thereby inhibiting cancer-associated thrombosis.
Antiplatelet drugs, including aspirin and clopidogrel, are commonly used for the secondary prevention of arterial thrombosis such as myocardial infarction and stroke. However, aspirin is not currently used for the prevention of ATE or VTE in patients with cancer; rather, it is used for chemoprevention of cancer, especially colorectal cancer. Recently, the US Preventive Services Task Force has suggested that many adults between 50 and 69 years of age would benefit from the use of aspirin for cancer prevention.16 Various mechanisms have been proposed for chemoprevention of colorectal cancer, including inactivation of platelets by inhibition of thromboxane A2 generation, as well as inactivation of epithelial cells by inhibition of WNT–β-catenin signaling through inhibition of prostaglandin E2 generation.17
Because VTE occurs more frequently than ATE in patients with cancer,2 relatively little attention has been paid to ATE. However, Navi et al18 have recently reported that the risk of arterial thromboembolic events begins to increase 150 days before the date of cancer diagnosis in older persons and peaks in the 30 days before cancer diagnosis. Although the detailed etiology has not been discussed, platelets may play a role in ATE in patients with cancer. A role of platelets will be discussed in the next section, focusing on a platelet activation receptor, C-type lectin-like receptor 2 (CLEC-2), and its endogenous ligand podoplanin.
Cancer cell–induced platelet aggregation and CLEC-2
Several kinds of cancer cells induce platelet aggregation and facilitate cancer growth and metastasis. Platelet aggregates the surrounding cancer cells to protect them from shear stress and/or natural killer cells in the blood and provide them with scaffolding for adhesion to the vascular wall. Moreover, angiogenic and growth factors secreted from activated platelets facilitate cancer growth and angiogenesis.19 Some cancer cells stimulate platelets through the expression of a membrane protein, podoplanin. Podoplanin is expressed on certain kinds of cancer cells, and its expression is reportedly associated with cancer metastasis or malignant progression.19 In 2007, we identified CLEC-2 as a receptor for podoplanin.20 Since then, it has been revealed that the association between CLEC-2 and podoplanin plays a large number of physiological and pathological roles.21
CLEC-2 was identified on platelets as a receptor for platelet-activating snake venom, rhodocytin.22 CLEC-2 protein is highly and almost specifically expressed in platelets and megakaryocytes and at lower levels in Kupffer cells in humans.21 In mice, very low expression of CLEC-2 has been reported in a small subset of dendritic cells.23 CLEC-2 mediates activation signals depending on protein tyrosine kinase. CLEC-2 has a signaling motif called “hemi-ITAM” (immunoreceptor tyrosine-based activation motif) in its cytoplasmic domain, which has a single YXXL motif. The clustering of CLEC-2 leads to tyrosine phosphorylation of hemi-ITAM by the Src kinases and the binding of the tyrosine kinase Syk to the tyrosine-phosphorylated hemi-ITAM. The activation of Syk induces downstream signaling events, including the tyrosine phosphorylation of SLP-76 and Syk and the activation of Bruton tyrosine kinase and phospholipase Cγ2 (PLCγ2), which culminate in platelet aggregation (Figure 1A).21,22
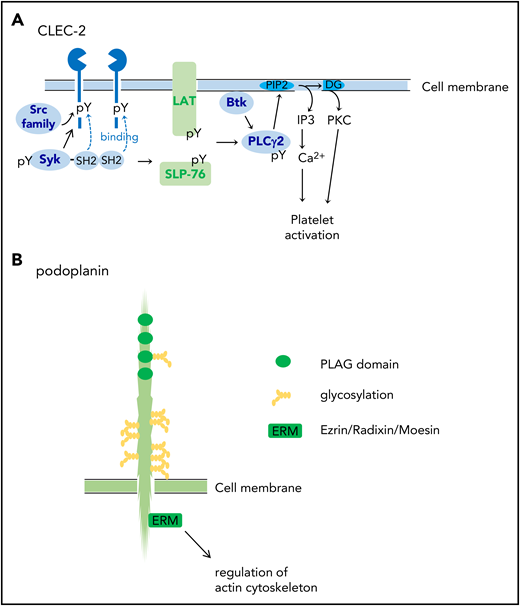
Structures of CLEC-2 and podoplanin. (A) CLEC-2 structure and its signal transduction pathway. CLEC-2 is cross-linked by podoplanin (endogenous ligand) or by rhodocytin (a platelet-activating snake venom) and undergoes tyrosine phosphorylation of hemi-ITAM (single YITL motif) by the Src family kinases. Then, the tyrosine kinase Syk binds to the phosphorylated ITAM motifs via its Src homology 2 (SH2) domains, which further increases Syk kinase activity. Syk phosphorylates adaptor proteins, LAT and SLP-76, which further activates downstream signaling molecules, including Bruton tyrosine kinase (Btk) and PLCγ2. PLCγ2 hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacylglycerol (DG) and inositol 1,4,5-trisphosphate (IP3), which leads to protein kinase C (PKC) activation and increase in Ca2+ mobilization from the open canalicular system (corresponding to endoplasmic reticulum), respectively. (B) Podoplanin structure. Podoplanin contains 4 platelet aggregation–stimulating (PLAG) domains, which are tandemly repeated and highly conserved. The podoplanin cytoplasmic domain is constitutively associated with ezrin/radixin/moesin (ERM) protein. Podoplanin clustering leads to functional effects through regulation of the actin cytoskeleton through ERM. pY, phosphorylated tyrosine.
Structures of CLEC-2 and podoplanin. (A) CLEC-2 structure and its signal transduction pathway. CLEC-2 is cross-linked by podoplanin (endogenous ligand) or by rhodocytin (a platelet-activating snake venom) and undergoes tyrosine phosphorylation of hemi-ITAM (single YITL motif) by the Src family kinases. Then, the tyrosine kinase Syk binds to the phosphorylated ITAM motifs via its Src homology 2 (SH2) domains, which further increases Syk kinase activity. Syk phosphorylates adaptor proteins, LAT and SLP-76, which further activates downstream signaling molecules, including Bruton tyrosine kinase (Btk) and PLCγ2. PLCγ2 hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacylglycerol (DG) and inositol 1,4,5-trisphosphate (IP3), which leads to protein kinase C (PKC) activation and increase in Ca2+ mobilization from the open canalicular system (corresponding to endoplasmic reticulum), respectively. (B) Podoplanin structure. Podoplanin contains 4 platelet aggregation–stimulating (PLAG) domains, which are tandemly repeated and highly conserved. The podoplanin cytoplasmic domain is constitutively associated with ezrin/radixin/moesin (ERM) protein. Podoplanin clustering leads to functional effects through regulation of the actin cytoskeleton through ERM. pY, phosphorylated tyrosine.
Podoplanin
Podoplanin is a sialomucin-like single transmembrane glycoprotein consisting of a short cytoplasmic domain and an extracellular domain with plenty of Ser/Thr residues as potential O-glycosylation sites. It is expressed on the surface of a wide range of cancer cells, including squamous cell carcinomas, seminomas, osteosarcoma, and glioblastoma.19,21 Podoplanin is also expressed in various kinds of normal cells, including podocytes in the kidney, type I alveolar cells in the lungs, fibroblastic reticular cells (FRCs) in the lymph nodes, synovial cells in the joint, and lymphatic endothelial cells (LECs), but not vascular endothelial cells.19,21 Podoplanin has four platelet aggregation–stimulating (PLAG) domains, which are tandemly repeated and highly conserved (Figure 1B). Among them, PLAG3 and PLAG4 domains, both of which contain Glu-Asp doublet and O-glycosylated Thr, are required for binding to CLEC-2 and podoplanin-dependent platelet aggregation.24,25 Experiments using Chinese hamster ovary (CHO) cell mutants lacking glycosylation pathways revealed that sialyl O-glycosylation of podoplanin is crucial for platelet aggregation–inducing activity.20
The podoplanin-mediated signaling pathway is largely unknown, but the podoplanin cytoplasmic tail binds ezrin/radixin/moesin (ERM) (Figure 1B), which interacts with both the plasma membrane and the actin filament.19 Podoplanin clustering by platelet CLEC-2 leads to the inhibition of LEC migration,26,27 stimulation of cell stretching,28,29 and epithelial–mesenchymal transition19 through ERM and regulation of the actin cytoskeleton.
CLEC-2–podoplanin interaction and cancer-related thrombosis
Podoplanin on the surface of cancer cells induces platelet aggregation, which facilitates hematogenous cancer metastasis. The pretreatment of CHO cells expressing human podoplanin with antipodoplanin blocking antibody NZ-1 significantly inhibited experimental lung metastasis in mice when NZ-1 was injected from the tail vein.19,24 Conversely, the injection of anti–CLEC-2 depleting antibody 2A2B10 into mice, followed by the injection of the podoplanin-expressing melanoma cell line B16F10, significantly inhibited the number of metastatic nodules in the lungs.30 We assume that blocking the interaction between CLEC-2 and podoplanin inhibits platelet adhesion to cancer cells, thereby shortening survival of cancer cells in the bloodstream.
Even in the absence of apparent metastasis to the distant organs, we observed that a substantial number of cancer cells are circulating in the bloodstream in cancer-bearing mice. We have reported that B16F10 orthotopically inoculated into the skin of mice did not spontaneously metastasize to the lung, but cancer cells circulating in the ear vessels were observed 21 days after B16F10 inoculation, the number of which was significantly reduced in CLEC-2–depleted mice.30 Therefore, podoplanin on the surface of cancer cells induces platelet aggregation in the bloodstream before the establishment of hematogenous distant metastasis. Indeed, Riedl et al31 evaluated the link between the expression of podoplanin in cancer and the prothrombotic state that is characteristic of glioma and found that podoplanin expression was related to altered markers of coagulation activation and an increased risk of VTE,31,32 although hematogenous metastasis of glioma is quite rare. Riedl et al31 suggested that podoplanin expressed on circulating glioma cells or glioma-derived microvesicles in the bloodstream activates platelets by binding to CLEC-2.
In our cancer-bearing mouse model, we observed massive thrombus formation in the lungs of the mice inoculated with podoplanin-positive B16F10 in the back skin, although spontaneous metastasis was not observed.30 The thrombus formation was greatly inhibited in the B16F10-bearing CLEC-2–depleted mice, suggesting that circulating podoplanin-positive B16F10 or microvesicles stimulate platelets and subsequent thrombus formation through CLEC-2–podoplanin interaction. However, podoplanin, which induces cancer-associated thrombosis, may not be restricted to podoplanin on the surface of cancer cells. Podoplanin is also expressed in normal tissues, including kidney podocytes, type I lung alveolar cells, FRCs in the lymph nodes, and LECs, but not in vascular endothelial cells. Podoplanin in these cells cannot have access to platelet CLEC-2. However, in pathological status, the ectopic podoplanin expression is induced. Payne et al33 reported that podoplanin expression is upregulated in the vessel wall during venous thrombosis, which may be induced by thromboinflammation.34 Moreover, Hitchcock et al35 reported that podoplanin expression is induced in macrophages or Kupffer cells in the liver upon Salmonella infection. These podoplanin-expressing cells migrate to the vicinity of vascular endothelial cells and interact with platelets leaking from hyperpermeable vessels, which facilitates thrombus formation in the liver. In addition, cancer-associated fibroblasts reportedly express podoplanin, which is correlated with cancer malignancy and poor prognosis in lung, breast, pancreatic, and liver cancer.19 This pathologically expressed podoplanin may also participate in cancer-associated thrombosis via interaction with platelet CLEC-2. The molecular mechanisms by which podoplanin induces cancer-associated thrombosis are summarized in Figure 2.
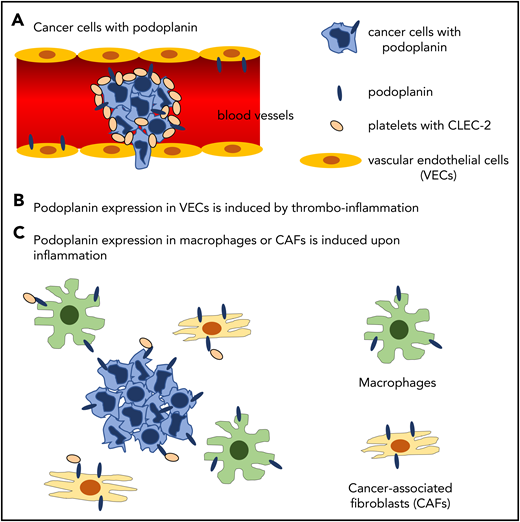
Hypothetical molecular mechanisms by which podoplanin induces cancer-associated thrombosis. (A) Podoplanin expressed on the surface of cancer cells activates platelets by binding to platelet CLEC-2 in the bloodstream. (B) Thromboinflammation induces expression of podoplanin in vascular endothelial cells, where podoplanin is not expressed in physiological status. (C) Podoplanin expression is induced in inflammatory macrophages and cancer-associated fibroblasts by inflammatory cytokines, although direct evidence shows that podoplanin in these cells activates platelets.
Hypothetical molecular mechanisms by which podoplanin induces cancer-associated thrombosis. (A) Podoplanin expressed on the surface of cancer cells activates platelets by binding to platelet CLEC-2 in the bloodstream. (B) Thromboinflammation induces expression of podoplanin in vascular endothelial cells, where podoplanin is not expressed in physiological status. (C) Podoplanin expression is induced in inflammatory macrophages and cancer-associated fibroblasts by inflammatory cytokines, although direct evidence shows that podoplanin in these cells activates platelets.
What is transcriptional regulation of podoplanin in physiological and pathological conditions? In normal LECs, podoplanin is regulated by the master regulator of lymphatic development, Prox-1.36 However, Prox-1 is not expressed in FRCs, suggesting that there are other regulators in FRCs. As for cancer cells, the major pathway of podoplanin upregulation in malignant conditions depends on the transcriptional factor activator protein (AP)-1. AP-1 acts through the phosphatidylinositol 3-kinase (PI3K)–Akt signaling pathway in skin cancers, osteosarcoma, and gliomas.37-40 Fos, one of the components of AP-1, directly binds to the podoplanin promoter and facilitates podoplanin expression.40 Podoplanin is also upregulated under inflammatory conditions. Its expression is induced by IL-1β, TNF-α, or transforming growth factor (TGF)-β1 in fibroblast-like synovial cells. Moreover, podoplanin is also upregulated in keratinocytes treated with TGF-β, IL-6, IL-22, or interferon-γ through Smad2/3/4 or STAT1/3. Inflammatory cytokines also induce podoplanin expression at the tumor invasive front of cervical squamous cell carcimona.41 Thus, podoplanin expression can be induced by different mechanisms, depending on conditions: Podoplanin in LECs is upregulated by Prox-1, whereas podoplanin in cancer cells and normal cells at inflammation sites is upregulated by the PI3K–Akt–AP-1 pathway and inflammatory cytokines.
We observed that cancer-bearing CLEC-2–depleted mice showed prolonged survival compared with cancer-bearing control mice, although distant metastasis was not observed, and the size of the inoculated cancer was the same in both groups.30 The cancer-bearing CLEC-2–depleted mice showed reduced plasma IL-6 and IL-1β, milder anemia, and less sarcopenia compared with cancer-bearing control mice. Because sarcopenia and anemia are symptoms of cancer cachexia, it may be possible that the activation of platelet CLEC-2 with podoplanin on the surface of cancer cells stimulates cancer-associated thrombosis, subsequent thromboinflammation, and inflammation-induced cachexia.
CLEC-2 and podoplanin as drug targets
The main cause of death in patients with cancer is tumor progression (cancer cachexia and distant metastasis), and the second leading cause is cancer-associated thrombosis.8 Therefore, drugs that block the association between CLEC-2 and podoplanin are good candidates for anticancer metastasis and anticancer-associated thrombosis treatments. In theory, these drugs would decrease hematogenous cancer metastasis, cancer-associated thrombus formation, thromboinflammation, and subsequent inflammation-induced cachexia.
Recently, Volz et al42 reported that the inhibition of platelet glycoprotein VI (GPVI) induces intratumoral hemorrhage and increases the efficacy of chemotherapy in mice. It has been reported that CLEC-2 and a collagen receptor GPVI are necessary for the maintenance of vascular integrity during inflammation but not physiological hemostasis.43 During inflammation by cancer, the absence of GPVI would lead to intratumoral hemorrhage and subsequent effective permeation of chemotherapeutic drugs. In the absence of CLEC-2, thrombus formation in the vessels of B16F10 inoculated into the back skin of the mice was greatly inhibited, although intratumoral hemorrhage was not increased.30 We would speculate that the efficacy of chemotherapy is also increased by CLEC-2 blockade because of the increase of functional vessels in cancer.
Because podoplanin is also expressed in normal tissues, the potential for adverse drug effects should be noted. To solve the problem, the LpMab monoclonal antibody series, which specifically recognizes aberrantly glycosylated podoplanin on cancer cells, has been reported.44 Although this antibody is a promising tool to avoid side effects, it may not recognize podoplanin upregulated in endothelial cells and/or monocytes during chronic inflammation, which would participate in cancer-related thrombosis.
Targeting CLEC-2 may have advantages in these viewpoints, because anti–CLEC-2 drugs would inhibit cancer-related thrombosis by blocking the association between CLEC-2 and podoplanin, both in cancer cells and in various normal cells, where podoplanin expression is pathologically induced. Moreover, CLEC-2 expression is highly restricted to platelets and megakaryocytes. The most probable side effect of anti–CLEC-2 drugs is increased bleeding tendency. However, mice that have been genetically and pharmacologically depleted of CLEC-2 do not show a significant increase in bleeding tendency,30,45,46 probably because the podoplanin expression is not observed in normal vessel walls and plays only a minimal role in physiological hemostasis. On the basis of our data derived from experiments in mice (data not shown), it was speculated that anti–CLEC-2 drugs would induce less bleeding tendency than that noted with antiplatelet/anticoagulation drugs. To date, anti–CLEC-2 antibodies,9,30 a CLEC-2-binding small molecule,47,48 and mutant recombinant rhodocytin49 have been reported as reagents to bind to CLEC-2 and block podoplanin binding. However, further study is required before clinical application.
Acknowledgments
The author thanks all the researchers who contributed to the studies on which this review is based, especially Toshiaki Shirai.
This work was supported in part by the Funding Program for Next Generation World-Leading Researchers (NEXT Program grant LS052).
Correspondence
Katsue Suzuki-Inoue, Department of Clinical and Laboratory Medicine, Faculty of Medicine, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi 409-3898, Japan; e-mail: katsuei@ yamanashi.ac.jp.
