
Abstract
Many drugs have been implicated in drug-induced immune thrombocytopenia (DITP). Patients with DITP develop a drop in platelet count 5 to 10 days after drug administration with an increased risk of hemorrhage. The diagnosis of DITP is often challenging, because most hospitalized patients are taking multiple medications and have comorbidities that can also cause thrombocytopenia. Specialized laboratory diagnostic tests have been developed and are helpful to confirm the diagnosis. Treatment of DITP involves discontinuation of the offending drug. The platelet count usually starts to recover after 4 or 5 half-lives of the responsible drug or drug metabolite. High doses of intravenous immunoglobulin can be given to patients with severe thrombocytopenia and bleeding. Although in most cases, DITP is associated with bleeding, life-threatening thromboembolic complications are common in patients with heparin-induced thrombocytopenia (HIT). Binding of antiplatelet factor 4/heparin antibodies to Fc receptors on platelets and monocytes causes intravascular cellular activation, leading to an intensely prothrombotic state in HIT. The clinical symptoms include a decrease in platelet counts by >50% and/or new thromboembolic complications. Two approaches can help to confirm or rule out HIT: assessment of the clinical presentation using scoring systems and in vitro demonstration of antiplatelet factor 4/heparin antibodies. The cornerstone of HIT management is immediate discontinuation of heparin when the disease is suspected and anticoagulation using nonheparin anticoagulant. In this review, we will provide an update on the pathophysiology, diagnosis, and management of both DITP and HIT.
Learning Objectives
Establish differential diagnosis
Develop appropriate upfront management for drug-associated thrombocytopenia
Introduction
Many therapeutic agents have been associated with thrombocytopenia. Although in most cases, drug-induced thrombocytopenia is associated with bleeding, life-threatening thromboembolic complications are common in patients with heparin-induced thrombocytopenia (HIT). The identification of the compound responsible for thrombocytopenia is often challenging, because most hospitalized patients are taking multiple medications and have comorbidities that can also cause thrombocytopenia. In this review, we will provide an update on the pathophysiology, diagnosis, and management of both drug-induced immune thrombocytopenia (DITP) and HIT.
Drug-mediated thrombocytopenia
In contrast to immune-mediated thrombocytopenia, nonimmune drug-induced thrombocytopenia is described as a direct cytotoxic effect of the drug molecules on the megakaryocytes and/or platelets, leading to dysfunctional thrombopoiesis within the bone marrow or increased platelet destruction in the circulation, respectively. Antineoplastic agents commonly cause thrombocytopenia, because many of these compounds are directly toxic to the hematopoietic stem cells. Myelosuppression was also shown to be a major treatment-related adverse event of the antibiotic linezolid.1 Although most antineoplastic agents are thought to mediate direct destruction of platelets or megakaryocytes, certain drugs of this group (for example, oxaliplatin) were found to cause acute, often severe thrombocytopenia mediated by drug-induced platelet antibodies.2,3
Certain drugs have been shown to directly mediate an antibody-independent platelet apoptosis by causing Ca+2 signaling, mitochondrial depolarization, and phosphatidylserine exposure in platelets (Table 1).4,5 Although these findings sound interesting, not all patients treated with these drugs experience some degree of thrombocytopenia. Hence, the clinical evidence that drug-induced apoptosis might be responsible for clinically significant thrombocytopenia is still missing, and future studies are needed to evaluate the effect of proapoptotic drug administration on platelet counts.
Drugs associated with nonimmune thrombocytopenia
| Suspected impaired thrombopoiesis | Suspected proapoptotic effect |
|---|---|
| Chemotherapy | Tamoxifen |
| Antineoplastics | Navitoclax |
| Interferon-a | Methotrexate |
| Linezolid | Nuclear factor–κB inhibitors |
| Botrezomib | Lovastatin |
| Thiazide diuretics | Doxorubicin |
| Ethanol | Bexarotene |
| Tolbutamid | Arsenic trioxide |
| Ganciclovir | Aspirin |
| Vancomycin | |
| Trifluoperazine | |
| Balhimycin | |
| Carmustine | |
| ABT-737 | |
| Cisplatin |
| Suspected impaired thrombopoiesis | Suspected proapoptotic effect |
|---|---|
| Chemotherapy | Tamoxifen |
| Antineoplastics | Navitoclax |
| Interferon-a | Methotrexate |
| Linezolid | Nuclear factor–κB inhibitors |
| Botrezomib | Lovastatin |
| Thiazide diuretics | Doxorubicin |
| Ethanol | Bexarotene |
| Tolbutamid | Arsenic trioxide |
| Ganciclovir | Aspirin |
| Vancomycin | |
| Trifluoperazine | |
| Balhimycin | |
| Carmustine | |
| ABT-737 | |
| Cisplatin |
DITP
More than 300 drugs have been implicated in DITP. A systematic review of individual patient data found that the most commonly reported drugs with a definite or probable causal relation to thrombocytopenia were quinine, quinidine, trimethoprim/sulfamethoxazole, vancomycin, penicillin, rifampin, carbamazepine, ceftriaxone, ibuprofen, mirtazapine, oxaliplatin, and suramin as well as the glycoprotein IIb/IIIa (GPIIb/IIa) inhibitors abciximab, tirofiban, and eptifibatide.6 The most common drug involved in DITP is, however, heparin.
The pathophysiology of the immune response in DITP
Several pathogenic mechanisms7 have been associated with DITP (Table 2). (1) Quinine-type drug-dependent antibodies (DDAbs). The classic DDAbs attach tightly to platelets only in the presence of the sensitizing drug and most often target GPIIb/IIIa or GPIb/IX. A recent study showed that a hybrid paratope consisting of quinine and reconfigured antibody, the complementarity-determining regions (CDRs) of the DDAbs, plays a critical role in recognition of its target epitope by an antibody.8 In other words, quinine binds directly to antibody CDRs, causing them to acquire specificity and avidity for a site on a platelet integrin. (2) Hapten-dependent DDAbs. small molecules (<5000 Da; eg, penicillin) require a covalent coupling to a larger carrier protein, mostly GPIIb/IIIa, to elicit drug-specific antibodies, which then bind to the small molecule drug rather than to the platelet protein. (3) Fiban-type DDAbs. Thrombocytopenia associated with use of fiban-type platelet inhibitors seems to be caused by antibodies that recognize immunogenic conformational changes induced in GPIIb/IIIa when the drug binds to the integrin.9 (4) Drug-specific DDAbs. Usually observed after administration of drugs with a murine component, such as abciximab, a chimeric (mouse-human) monoclonal antibody Fab fragment specific for GPIIIa is used primarily to prevent platelet aggregate formation. Drug-specific antibodies that seem to recognize murine sequences CDR3 of abciximab are responsible for this type of DITP. (5) Autoantibody mechanism. These antibodies are induced after drug exposure (especially gold therapy) but are not dependent on the presence of the drug for their binding to platelets. (6) Immune complexes. Some DDAbs form immune complexes with their antigens. These complexes are able to activate platelets via the Fcγ receptors. This mechanism will be discussed in detail below.
Mechanisms of DITP
| Type of antibody | Mechanism | Example of drugs |
|---|---|---|
| Quinine type | Drug binds DDAbs and subsequently, platelet integrin | Quinine, sulfonamide antibiotics, nonsteroidal anti-inflammatory drugs |
| Hapten dependent | Drug links covalently to membrane protein and induces drug-specific binding by DDAbs | Penicillin, some cephalosporin antibiotics |
| Fiban type | Drug reacts with glycoprotein IIb/IIIa and induces neoepitope(s) for the DDAbs | Tirofiban, eptifibatide |
| Drug specific | DDAbs recognize the murine component of chimeric Fab fragment specific for glycoprotein IIIa | Abciximab |
| Autoantibody | Drug induces antibody that reacts with autologous platelets in the absence of drug | Gold salts, procainamide |
| Immune complexes | Antibodies form immune complexes with their target antigens | Heparin, protamine |
| Type of antibody | Mechanism | Example of drugs |
|---|---|---|
| Quinine type | Drug binds DDAbs and subsequently, platelet integrin | Quinine, sulfonamide antibiotics, nonsteroidal anti-inflammatory drugs |
| Hapten dependent | Drug links covalently to membrane protein and induces drug-specific binding by DDAbs | Penicillin, some cephalosporin antibiotics |
| Fiban type | Drug reacts with glycoprotein IIb/IIIa and induces neoepitope(s) for the DDAbs | Tirofiban, eptifibatide |
| Drug specific | DDAbs recognize the murine component of chimeric Fab fragment specific for glycoprotein IIIa | Abciximab |
| Autoantibody | Drug induces antibody that reacts with autologous platelets in the absence of drug | Gold salts, procainamide |
| Immune complexes | Antibodies form immune complexes with their target antigens | Heparin, protamine |
DDAb, drug-dependent antibody.
Clinical features of DITP
DITP is a life-threatening clinical syndrome that is associated with a high risk of hemorrhage. A review of 247 case reports of DITP found incidence rates of major and fatal bleeding of 9% and 0.8%, respectively. Thrombocytopenia characteristically occurs ∼5 to 10 days after initial drug exposure, with median nadir platelet counts of <20 × 109/L. An exception is thrombocytopenia induced by the GPIIb/IIIa antagonists, which may present within hours of exposure (early onset) due to naturally occurring antibodies.
How do I diagnose DITP?
Diagnosis of DITP usually requires a high grade of clinical suspicion and a careful “detective” workup to identify the causative drug. Five clinical criteria can help to establish the diagnosis of DITP6,10 : (1) exposure to the candidate drug was preceded thrombocytopenia; (2) recovery from thrombocytopenia was complete and sustained after discontinuing the candidate drug; (3) the candidate drug was the only drug used before the onset of thrombocytopenia, or other drugs were continued or reintroduced after discontinuation of the candidate drug with a sustained normal platelet count; (4) other causes for thrombocytopenia were excluded; and (5) reexposure to the candidate drug resulted in recurrent thrombocytopenia even if this criterion is not applicable to HIT due to the lack of antigen‐specific memory B cells.11,12 However, because DITP often occurs in hospitalized patients who are taking multiple medications and have comorbidities that can also cause thrombocytopenia, relating thrombocytopenia to a particular drug depending solely on clinical information is difficult. Therefore, most investigators agree that confirmation requires either a drug challenge or the demonstration of DDAbs in vitro. Specialized laboratory testing for antibodies that bind to platelets in the presence of drugs or a drug metabolite has been developed. It provides useful confirmation of DITP. Test methods, which are mostly flow cytometry based, must show drug dependence, immunoglobulin binding, and platelet specificity, and ideally, they should be reproducible across laboratories. Recently, recommendations for laboratory testing for DITP have been published, in which the authors provided helpful methodological guidelines to increase the specificity and sensitivity of the used assays.13 Nevertheless, we have to take into account that negative test results are often obtained in patients with a clinical history strongly suggestive of DITP, which makes the diagnosis more complicated. One possible reason is that the available methods are not sufficiently sensitive. Another is that many antibodies may be specific for drug metabolites and may not be detected unless the correct metabolite is used in testing.
Management of patients suspected of having DITP
Treatment of DITP involves discontinuation of the offending drug. In cases of multiple medications, all drugs started within the last 2 weeks should be stopped (especially antibiotics) and replaced if necessary. The platelet count usually starts to recover after 4 to 5 half-lives of the responsible drug or drug metabolite. Of note, platelet transfusion is generally ineffective as long as the drug or its metabolites are present in plasma. High doses of intravenous immunoglobulin (IVIG) can be given to patients with severe thrombocytopenia and bleeding as well as those at high risk of bleeding; however, this recommendation is based only on case reports.14,15 Figure 1 provides a suggested approach to the management of cases with suspected DITP.
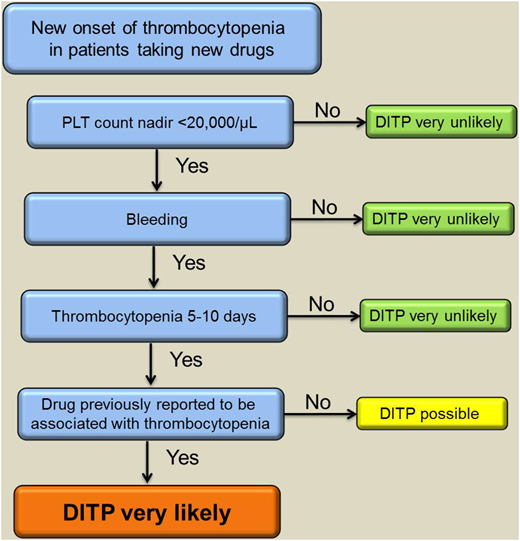
A suggested algorithm to verify the diagnosis of DITP based on clinical assessment supported by complementary laboratory investigations. PLT, platelet. Adapted from Transfus Med Rev., 27(3), Arnold DM, Nazi I, Warkentin TE, et al. Approach to the diagnosis and management of drug-induced immune thrombocytopenia, 137-145, Copyright (2013), with permission from Elsevier.
A suggested algorithm to verify the diagnosis of DITP based on clinical assessment supported by complementary laboratory investigations. PLT, platelet. Adapted from Transfus Med Rev., 27(3), Arnold DM, Nazi I, Warkentin TE, et al. Approach to the diagnosis and management of drug-induced immune thrombocytopenia, 137-145, Copyright (2013), with permission from Elsevier.
HIT
Nonimmune heparin-associated thrombocytopenia (formerly HIT type 1)
Thrombocytopenia can occur in 10% to 30% of patients treated with heparin in the absence of an obvious involvement of the immune system. The underlying pathophysiology is thought to be mediated by direct binding of heparin to platelets, resulting in mild platelet activation. In fact, it has been shown that binding of unfractionated heparin (UFH), low–molecular weight heparin (LMWH), or fondaparinux to the GPIIb/IIIa complex potentiates outside-in signaling in platelets.16
In nonimmune heparin-associated thrombocytopenia, thrombocytopenia, which typically occurs within the first few days (earlier than day 5) of heparin therapy, is usually mild and without major clinical consequence. Platelet count often remains above 80 to 100 × 109/L, and it spontaneously recovers to baseline levels in a few days despite continuous heparin treatment. Patients do not experience any bleeding or thrombotic complications and do not require initiation of therapy.
Immune HIT (HIT type 2)
Immune-mediated HIT is a prothrombotic disorder that occurs after exposure to UFH or LMWH.17 When injected, heparin reacts with platelet factor 4 (PF4) to produce immunogenic complexes that induce antibodies specific for PF4 in a complex with heparin or other macromolecular polyanions. Only a minority of the immunized patients, however, develop HIT characterized by a fall in platelet count beginning between days 5 and 10 of heparin therapy with or without thromboembolic events.18 A 10-fold lower risk of HIT is observed with use of a prophylactic dose of LMWH compared with UFH. One recent study showed a dramatic reduction in the number of cases with suspected HIT after the implementation of a strategy to avoid UFH and replace it with LMWH if required.19
Pathophysiology of HIT.
PF4 binds in a charge-dependent fashion to gram-positive and gram-negative bacteria. In a mouse model of polymicrobial bacterial sepsis, anti-PF4/heparin-reactive antibodies were generated in the absence of heparin exposure. Interestingly, immunoglobulin G (IgG) anti-PF4/heparin antibodies are frequently found even in heparin-naïve individuals, and an association has been reported between bacterial periodontal disease and anti-PF4/heparin antibodies.20 These observations suggest that immune response against PF4/polyions complexes is an ancient host defense mechanism and that HIT is simply the consequence of a misdirected immune response to heparin-induced epitopes on PF4.17
Not all antibodies against PF4/heparin complexes are capable of platelet activation and inducing clinical HIT. Only a subset of these antibodies, linked to the PF4/heparin complexes, binds with their Fc parts to FcγRIIA receptors on platelets. Crosslinking of the Fc receptors leads to platelet activation. This results in release of platelet granules, formation of platelet microparticles, thrombin generation, and ultimately, platelet aggregation.17 Endothelium and monocyte activation with tissue factor expression is also involved in the pathophysiology of HIT.21 Recent studies show that pathogenic HIT antibodies bind to PF4-coated monocytes and activate them via FcγRIIA, leading to expression of tissue factor and generation of thrombin.22 These processes are thought to be responsible for the hypercoagulable state of HIT and the frequent occurrence of thrombotic complications in the absence of anticoagulation. Although no significant impact of traditional thrombophilic markers (eg, factor V Leiden) was found on the risk of HIT, one recent study reported on a higher risk of thrombosis in individuals homozygous for the 131-RR genotype of the FcγRIIA.23 The authors provided evidence on the inability of endogenous monomeric IgG2 in those individuals to effectively compete with HIT immune complexes for binding platelet FcγRIIA.
Clinical manifestations of HIT.
Patients with HIT can present with a wide spectrum of symptoms. The cardinal clinical feature is a fall in platelet count >50% (from the highest value after the start of heparin treatment) typically beginning 5 to 10 days after starting heparin therapy. HIT can also manifest rapidly in patients who have received heparin in the previous 100 days (rapid-onset HIT). Although a mean nadir between 50 and 80 × 109/L was most often found in larger cohort studies, HIT cases that are complicated by disseminated intravascular coagulation (DIC) may result in a deeper drop in platelet count <20 × 109/L.
Other than thrombocytopenia, HIT may also be associated with thrombosis, which is the most severe complication of HIT and contributes to disease morbidity and mortality. About 1/2 of untreated patients with acute HIT develop a new thrombotic complication. Deep vein thrombosis, with or without pulmonary embolism, is the most common complication. Less common is concurrent or recent intravascular catheter use thrombosis in cerebral and splanchnic veins. Arterial thrombosis is less frequent than venous thrombosis in HIT patients and typically involves lower-limb, cerebral, coronary, mesenteric, and brachial arteries.17 Rarely, severe HIT-associated DIC leads to microthrombosis and critical limb ischemia, even in the absence of warfarin therapy. Other (rare) complications observed in HIT patients include skin necrosis at the heparin injection sites and adrenal hemorrhagic necrosis.
Autoimmne HIT.
This severe form of HIT may present as delayed-onset HIT, persisting HIT, spontaneous HIT syndrome, fondaparinux-associated HIT, heparin “flush”–induced HIT, and HIT-induced DIC.24 Sera from patients with autoimmune HIT contain antibodies that are able to activate platelets even in the absence of heparin. Using biomechanical experimental settings, it has been shown that these antibodies are able to bridge 2 PF4 tetramers even in the absence of heparin, leading to the formation of large multimolecular immune complexes and marked platelet activation.25 These findings may explain the persistence of thrombocytopenia for several weeks in patients with autoimmune HIT despite heparin discontinuation and why standard anticoagulant HIT therapy seems to be less effective. In contrast, a recent case report indicates that high-dose intravenous immunoglobulin can accelerate platelet count recovery.15
Identifying the pretest risk of HIT
To assist clinicians in this process, several clinical scoring systems for HIT have been developed. The most extensively studied scoring system, the 4Ts, incorporates 4 typical clinical features of HIT: (1) thrombocytopenia, (2) timing of onset of thrombocytopenia, (3) thrombosis or other clinical sequelae, and (4) the likelihood of other causes of thrombocytopenia (Table 3).26 The 4Ts system is helpful in identifying patients who are unlikely to have HIT.27 The negative predictive probability of a 4Ts score <4 has been shown to be very high (99.8%; 95% confidence interval, 97%-100%). However, the positive predictive values of an intermediate or even high 4Ts score are unsatisfactory (14%; 95% confidence interval, 9%-22% and 64%; 95% confidence interval, 40%-82%, respectively).28 Other scoring systems, such as the HIT expert probability score and the Lillo–Le Louët score, require more validation in prospective studies before a firm conclusion can be drawn on their performance in the diagnostic workup of HIT.
The 4Ts scoring system to evaluate the pretest risk for HIT
| 4Ts | 2 points | 1 point | 0 point |
|---|---|---|---|
| Thrombocytopenia | Platelet count fall >50% and platelet nadir ≥20 × 109/L | Platelet count fall 30%-50% or platelet nadir 10-19 × 109/L | Platelet count fall <30% or platelet nadir <10 × 109/L |
| Timing of platelet count fall | Clear onset between days 5 and 10 or platelet fall ≤1 d* | Onset after day 10 or fall ≤1 d† | Platelet count fall <4 or >14 d after exposure |
| Thrombosis or other sequelae | New thrombosis‡ (confirmed) | Suspected thrombosis (not proven) | None |
| Other causes for thrombocytopenia | None apparent | Possible | Definite |
| 4Ts | 2 points | 1 point | 0 point |
|---|---|---|---|
| Thrombocytopenia | Platelet count fall >50% and platelet nadir ≥20 × 109/L | Platelet count fall 30%-50% or platelet nadir 10-19 × 109/L | Platelet count fall <30% or platelet nadir <10 × 109/L |
| Timing of platelet count fall | Clear onset between days 5 and 10 or platelet fall ≤1 d* | Onset after day 10 or fall ≤1 d† | Platelet count fall <4 or >14 d after exposure |
| Thrombosis or other sequelae | New thrombosis‡ (confirmed) | Suspected thrombosis (not proven) | None |
| Other causes for thrombocytopenia | None apparent | Possible | Definite |
The resulting clinical probability score is divided into high (6-8 points), intermediate (4-5 points), and low (≤3 points).
In case of prior heparin exposure during the last 30 days.
In case of prior heparin exposure within 30 to 100 days.
Also skin necrosis, acute systemic reaction postintravenous UFH bolus, progressive or recurrent thrombosis, and non-necrotizing (erythematous) skin lesions.
Laboratory investigations
Two classes of assays are available: functional (platelet activation) assays and (PF4-dependent) immunoassays.29,30 The presence of platelet-activating antibodies can be established only using functional assays. Although recent studies indicated the feasibility of detection of platelet-activating antibodies using the whole-blood impedance analyzer,31 assays using washed platelets, such as the heparin-induced platelet activation (HIPA) assay and the serotinin release assay (SRA), are the gold standard in the laboratory diagnosis of HIT.32,33 Functional assays combine both high sensitivity and specificity for clinically relevant HIT antibodies. The sensitivity of SRA is currently under debate. Although the test was recently shown to be able to detect platelet-activating anti-PF4/heparin antibodies at the earliest onset of thrombocytopenia in HIT patients,34 other studies suggested a possible improvement of the ability to detect pathogenic HIT antibodies by adding exogenous PF4 before or during SRA.35,36
Although both functional assays are considered the “gold standard” for diagnosing HIT, these assays are difficult to perform, require selected healthy platelet donors, and are restricted to a few reference laboratories. A recent study showed that platelet-activating antibodies can be detected by flow cytometer.35 Using the PF4-dependent P-selectin expression assay, the authors showed in a follow-up study that the addition of PF4 enabled detection of pathogenic antibodies before the SRA became positive in 2 patients with HIT.37
Antibody binding can be detected by enzyme-linked immunosorbent assays (ELISA) and particle-based immunoassays. Although ELISAs have an excellent negative predictive value to rule out HIT, their specificity is low (40%-80%).29 Several approaches may increase the diagnostic specificity of ELISAs. These include exclusive detection of anti-PF4/heparin IgG antibodies, consideration of the magnitude of the optical density value, and implementation of a confirmative inhibition step. Recent meta-analysis, however, did not find a significant advantage of IgG-specific ELISAs over polyspecific ELISAs to improve the overall performance characteristics of the immunoassays.38 Particle-based immunoassays are easily performed, and reactions can be detected either visually after centrifugation as in the particle gel immunoassay or using lateral flow technology. The major advantage of these assays is the rapid turnaround time. Recent studies showed high negative predictive values of these assays.39 Automated particle-based immunoassays have also been introduced.40 A systematic meta-analysis recently investigated the diagnostic accuracy of rapid immunoassays for HIT. Data from this study showed that rapid immunoassays for HIT have high negative predictive value and can be used to exclude HIT, particularly in patients with low or intermediate clinical probability.41 The relative high costs are still, however, an issue for small-sized laboratories.
Management of patients with suspected HIT
Given that HIT is an immune reaction enhanced by heparin, it is mandatory to discontinue all heparins when HIT is strongly suspected. However, due to the partly autoimmune nature of HIT, discontinuation of heparin alone is not adequate. Therefore, patients with high clinical suspicion of HIT should be promptly treated with a nonheparin anticoagulant while awaiting laboratory confirmation or exclusion of the diagnosis. Different anticoagulants are currently used to treat patients with HIT. However, alternative anticoagulants are rarely used outside the niche indication of HIT, and many physicians have limited experience handling these drugs. This may increase the risk for both bleeding and thrombotic events. Therefore, it is extremely important that clinicians are able to distinguish between patients who actually have HIT and the more common patients who may have PF4/heparin-specific antibodies and some degree of thrombocytopenia but do not have HIT. Diagnostic algorithms that combine clinical features and results of laboratory testing are available for diagnosis of HIT. A suggested example is shown in Figure 2.
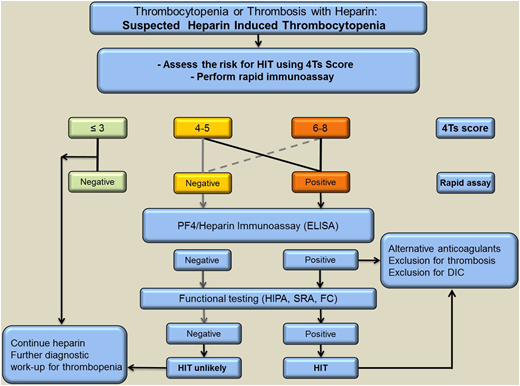
A suggested approach to diagnosis and initial management of patients with suspected HIT based on clinical assessment supported by complementary laboratory investigations. Screening PF4-dependent immunoassays is indicated for patients with at least intermediate probability of HIT. If the ELISA is positive, a functional assay should also be performed to confirm or refute a diagnosis of HIT. FC, flow cytometer.
A suggested approach to diagnosis and initial management of patients with suspected HIT based on clinical assessment supported by complementary laboratory investigations. Screening PF4-dependent immunoassays is indicated for patients with at least intermediate probability of HIT. If the ELISA is positive, a functional assay should also be performed to confirm or refute a diagnosis of HIT. FC, flow cytometer.
Alternative anticoagulants for HIT
Parenteral anticoagulants
Activated factor X inhibitors.
In a prospective, randomized trial, danaparoid was shown to be efficient in preventing new, progressive, or recurrent thromboembolic complications (including thrombotic death) or limb amputation in HIT.42 This seems to be mediated by (1) low crossreactivity rate with HIT antibodies in vitro and in vivo, (2) the unique property of specific suppression of HIT antibody–induced platelet activation by replacing PF4/heparin complexes from the platelet surface, and (3) disruption of PF4/heparin complexes.
Fondaparinux is a synthetic pentasaccharide with potent indirect anti-Xa inhibitor properties that have been increasingly used off label for the management of HIT.43 Fondaparinux was found to be safe for patients with acute thrombosis with heparin-dependent platelet-activating antibodies.44 Another study also showed similar effectiveness and safety as argatroban and danaparoid in patients with suspected HIT treated with fondaparinux.45
Direct thrombin inhibitors.
Argatroban is a synthetic direct thrombin inhibitor that reversibly binds to the thrombin active site. It is capable of inhibiting both free and clot-associated thrombin. Two multicenter trials showed that argatroban therapy reduces death, amputation, and thrombosis compared with historical controls.46
Bivalirudin is another synthetic peptide composed of 2 short hirudin peptide fragments. It is the best investigated alternative anticoagulant in non-HIT patients with coronary disease, including acute coronary syndrome and that requiring coronary intervention.47
Direct oral anticoagulants
Rivaroxaban, apixaban, and endoxaban directly inhibit activated factor X, whereas dabigatran is a direct thrombin inhibitor. Emerging evidence suggests the safety and efficacy of several direct oral anticoagulants in HIT. In a small multicenter, prospective study, rivaroxaban seemed to be safe and effective without occurrence of new thrombosis.48 In another case series, patients with HIT who were treated with rivaroxaban, apixaban, or dabigatran had no recurrent arterial or venous thromboses or bleeding complications.49-54 Although these observations sound promising, published experience with these drugs in patients with acute HIT is limited and does not allow for final conclusions on their safety and efficacy. Of particular importance seems to be the observed low trough levels of the drug, which might cause inadequate protection for HIT patients.
High doses of IVIG were shown to inhibit HIT antibody-mediated platelet activation. Accumulating evidence from case reports suggests that patients with prolonged thrombocytopenia refractory to standard treatment may benefit from IVIG therapy.55
Choice and duration of the anticoagulation
The decision of which nonheparin anticoagulant to use should be based on the patient’s clinical stability, hepatic and renal function, and most importantly, physician’s expertise. Several advantages and disadvantages of nonheparin anticoagulants should be taken into consideration when selecting an agent (Table 4).
Nonheparin alternative anticoagulants that may be used in HIT patients
| Anticoagulant | Mechanism of action | Application | Clearance | Half-life time | Monitoring |
|---|---|---|---|---|---|
| Parenterale | |||||
| Argatroban | Direct thrombin inhibition | Intravenously | Hepatic | 40-50 min | aPTT |
| Bivalirudin | Direct thrombin inhibition | Intravenously | Renal | 25 min | aPTT |
| Danaparoid | Indirect inhibition of FXa | Intravenously, subcutaneously | Renal | 24 h | Danapariod–anti-Xa |
| Fondaparinux | Indirect inhibition of FXa | Subcutaneously | Renal | 17-24 h | Fondaparinux–anti-Xa |
| Oral | |||||
| Dabigatran | Direct thrombin inhibition | Per oral | Renal (∼85%) | 12-14 h | — |
| Rivaroxaban | Direct inhibition of FXa | Per oral | Renal (∼33%) | 5-9 h | — |
| Apixaban | Direct inhibition of FXa | Per oral | Renal (∼25%) | 8-15 h | — |
| Endoxaban | Direct inhibition of FXa | Per oral | Renal (∼50%) | 10-14 h | — |
| Anticoagulant | Mechanism of action | Application | Clearance | Half-life time | Monitoring |
|---|---|---|---|---|---|
| Parenterale | |||||
| Argatroban | Direct thrombin inhibition | Intravenously | Hepatic | 40-50 min | aPTT |
| Bivalirudin | Direct thrombin inhibition | Intravenously | Renal | 25 min | aPTT |
| Danaparoid | Indirect inhibition of FXa | Intravenously, subcutaneously | Renal | 24 h | Danapariod–anti-Xa |
| Fondaparinux | Indirect inhibition of FXa | Subcutaneously | Renal | 17-24 h | Fondaparinux–anti-Xa |
| Oral | |||||
| Dabigatran | Direct thrombin inhibition | Per oral | Renal (∼85%) | 12-14 h | — |
| Rivaroxaban | Direct inhibition of FXa | Per oral | Renal (∼33%) | 5-9 h | — |
| Apixaban | Direct inhibition of FXa | Per oral | Renal (∼25%) | 8-15 h | — |
| Endoxaban | Direct inhibition of FXa | Per oral | Renal (∼50%) | 10-14 h | — |
aPTT, activated partial thromboplastin time; FXa, activated coagulation factor X.
Protamine/HIT
Protamine is widely used in medicine as an additive to certain preparations of insulin (delaying onset and prolonging duration of insulin action) and as a rapidly acting antidote to heparin, particularly to neutralize the effects of high heparin concentrations needed for anticoagulation during cardiac surgical procedures. Protamine and heparin form multimolecular complexes, which result in high rates of immunization in postcardiac surgery patients.56-59 A subset of antiprotamine/heparin IgGs activates platelets through their FcγIIA receptors and is thought to be associated with side effects, in particular thrombocytopenia.
Diagnosis of protamine/HIT
Antiprotamine/heparin antibodies can be detected using ELISAs. Heparin has been shown to increase binding of antiprotamine antibodies compared with protamine alone.60,61 In fact, protamine undergoes conformational changes after complexing with heparin,56,57 making it very likely that these antibodies bind to neoepitopes expressed on protamine only after complex formation with heparin. The ability of antiprotamine/heparin antibodies to activate platelets can be investigated in vitro using different laboratory methods, including SRA and HIPA assay.56,58,59,62,63 In a recently developed flow cytometer–based assay, we observed that the ratio between protamine and heparin is very critical for platelet activation.64
Clinical presentation of antiprotamine/heparin antibodies
Although the presence of antiprotamine/heparin antibodies (IgG/A/M) reportedly had no overall impact on the postoperative platelet count evolution,59 platelet-activating antibodies against protamine/heparin complexes before surgery have been shown to be associated with lower postoperative platelet counts and require longer times to return to the same (or greater) platelet count observed before surgery.56,63 The association between thrombocytopenia and platelet-activating antiprotamine/heparin antibodies has also been reported in several case reports.60,62 In addition, thromboembolic complications have been reported in patients with platelet-activating antibodies against protamine/heparin complexes,60,65 supporting evidence from some cohort studies.56,63 Of note, one recent case series reported on the use of argatroban in 4 patients with protamine/HIT. Platelet count recovered after starting argatroban, and no adverse events occurred.65
Conclusion
Thrombocytopenia after drug administration can be associated with bleeding or thrombosis depending of the pathophysiology of platelet destruction. Significant progress has been made during the last 2 decades in understanding the pathomechanisms of drug-associated thrombocytopenia. However, there are still numerous diagnostic and treatment challenges, especially in the critically ill patient, including the difficulty in distinguishing drug-associated thrombocytopenia from secondary thrombocytopenia caused by underlying disorders, like sepsis or tumor.
Current diagnostic test methods seem to have limited clinical added benefit in the management of patients suspected to have drug-associated thrombocytopenia. These assays are not automated, they are time consuming, and they require high technical expertise. This makes them restricted to reference laboratories. Thus, in many cases, the diagnosis is made based on clinical features without laboratory conformation. In addition, the sensitivity of the serological testing for DDAbs is generally considered to be low. In fact, negative test results are often obtained in patients with a clinical picture strongly suggestive of DITP. One possible reason is that available methods are not sufficiently sensitive due to the low avidity of DDAbs or the lack of the solubility of the target drug. Another is that many antibodies may be specific for drug metabolites and may not be detected unless the correct metabolite is used in testing. Finally, the mechanism of thrombocytopenia could simply not be related to platelet destruction but rather, could be inhibited platelet production. The implementation of megakaryocytes as test cells to investigate the binding of DDAbs and the impact of proplatelet production might improve the test sensitivity in DITP. In the absence of sensitive and easy-to-perform assays for DDAbs, physicians should stop drugs that are very likely responsible for thrombocytopenia, even if laboratory assays revealed negative test results.
In contrast to DITP, most laboratory investigations for HIT lack the specificity. Immunoassays that detect the binding of anti-PF4/heparin complexes have high sensitivity but unsatisfactory specificity, making confirmative testing using functional assays, such as HIPA or SRA, indispensable, particularly in patients who tested positive in the immunoassay. Recently, important efforts have been made to improve the performance of the functional assays, including addition of PF4 to increase the sensitivity of SRA or flow cytometer-based assays. Although the results of these studies are promising, additional validation in multicenter laboratory workshops as well as clinical trials is needed before a final conclusion could be drawn.
A better understanding of pathophysiology of the different drug-associated thrombocytopenias may help developing strategies to avoid complications induced by these drugs. For instance, some drugs are able to induce conformational changes in platelet surface proteins, leading to increased binding avidity of DDAbs. Using new techniques, like circular dichroism spectroscopy and atomic force microscopy, to analyze protein changes in the presence of the drug could be helpful to predict the immunization risk in treated patients.
Acknowledgment
The authors thank Stephen Bosher and Karina Althaus for helpful discussion.
Correspondence
Tamam Bakchoul, Medical Faculty of Tubingen, Otfried-Müller-Strasse 4/1, 72076 Tubingen, Germany; e-mail: tamam.bakchoul@med.uni-tuebingen.de.
