
Abstract
Significant advances have been made in diagnosis and clinical management of inherited red cell membrane disorders that result in hemolytic anemia. Membrane structural defects lead to hereditary spherocytosis (HS) and hereditary elliptocytosis (HE), whereas altered membrane transport function accounts for hereditary xerocytosis (HX) and hereditary overhydrated stomatocytosis (OHS). The degrees of membrane loss and resultant increases in cell sphericity determine the severity of anemia in HS and HE, and splenectomy leads to amelioration of anemia by increasing the circulatory red cell life span. Alterations in cell volume as a result of disordered membrane cation permeability account for reduced life span red cells in HX and OHS. Importantly, splenectomy is not beneficial in these 2 membrane transport disorders and is not recommended because it is ineffective and may lead to an increased risk of life-threatening thrombosis. Rational approaches are now available for the diagnosis and management of these inherited red cell disorders, and these will be discussed in this review.
Learning Objectives
Understand the cellular basis for altered red cell function and resultant hemolytic anemia in inherited membrane disorders that are the result of membrane structural defects or as a consequence of altered cell volume regulation
Provide strategies for recognizing and diagnosing these disorders and clinical management of the resultant hemolytic anemia
Intrinsic erythrocyte membrane disorders
Besides hemoglobinopathies and enzymopathies, intrinsic erythrocyte membrane disorders are a common cause of hemolytic anemia in children and adults. This review discusses pathophysiology, inheritance, clinical findings, laboratory and diagnostic features, and current management practices for 4 common and well-studied inherited red cell membrane disorders: spherocytosis, elliptocytosis, xerocytosis, and overhydrated stomatocytosis (OHS).
Significant progress has been made in defining the detailed structural organization of the red cell membrane (Figure 1), which plays a critical role in enabling the human red cells to undergo extensive, rapid, and reversible deformations as they pass through the microvasculature and perform their function of delivering oxygen and transiting the human spleen.1 The membrane is a composite structure of a lipid bilayer anchored to a two-dimensional elastic network of skeletal proteins through ankyrin- and protein 4.1–based transmembrane protein complexes embedded in the lipid bilayer. The vertical anchoring linkages provide cohesion between the lipid bilayer and the membrane skeleton. Loss of cohesion between the lipid bilayer and the membrane skeleton leads to the loss of membrane surface area. The horizontal linkages between spectrin-spectrin dimers and between spectrin-actin-protein 4.1R in the junctional complex regulate membrane deformability and membrane mechanical integrity. Defective horizontal linkages lead to decreased membrane mechanical integrity and cell fragmentation. The spleen is the quality-control organ for red cell function in that it sequesters cells with reduced cellular deformability and removes them from the circulation.2
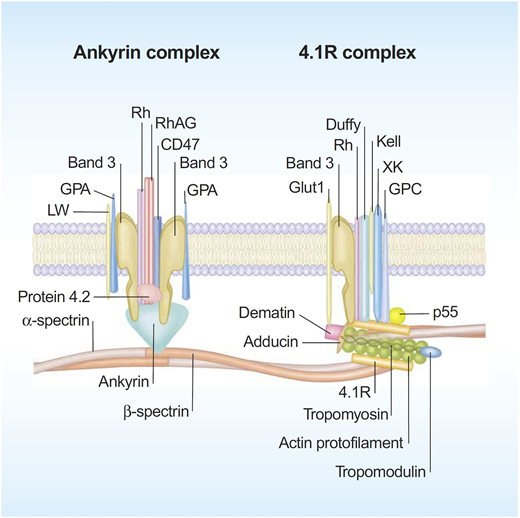
A schematic representation of the human red cell membrane. The membrane is a composite structure of a lipid bilayer anchored to a two-dimensional elastic network of skeletal proteins through ankyrin- and protein 4.1–based transmembrane protein complexes embedded in the lipid bilayer. Deficiency in any of the proteins involved in the vertical linkages involving the ankyrin complex leads to loss of cohesion between the lipid bilayer and membrane skeleton and the resultant loss of membrane surface area. The horizontal linkages between spectrin-spectrin dimers and between spectrin-actin-protein 4.1R in the junctional complex determine red cell membrane mechanical integrity. Defective horizontal linkages lead to decreased integrity of the membrane mechanics and cell fragmentation. Reproduced with permission from Mohandas and Gallagher.
A schematic representation of the human red cell membrane. The membrane is a composite structure of a lipid bilayer anchored to a two-dimensional elastic network of skeletal proteins through ankyrin- and protein 4.1–based transmembrane protein complexes embedded in the lipid bilayer. Deficiency in any of the proteins involved in the vertical linkages involving the ankyrin complex leads to loss of cohesion between the lipid bilayer and membrane skeleton and the resultant loss of membrane surface area. The horizontal linkages between spectrin-spectrin dimers and between spectrin-actin-protein 4.1R in the junctional complex determine red cell membrane mechanical integrity. Defective horizontal linkages lead to decreased integrity of the membrane mechanics and cell fragmentation. Reproduced with permission from Mohandas and Gallagher.
Hereditary spherocytosis
Pathophysiology and inheritance
Hereditary spherocytosis (HS) is the most commonly inherited membrane disorder resulting from the assembly of a structurally dysfunctional red cell membrane.3,4 Progressive loss of membrane surface area due to decreased cohesion of the lipid bilayer to the spectrin-based membrane skeleton results in the generation of progressively more spherical cells with reduced cellular deformability that are sequestered in the spleen with resultant hemolytic anemia. HS occurs in all racial groups.
Inheritance of dominant, recessive, or de novo mutations in genes encoding red cell membrane proteins, ankyrin, band 3, α-spectrin, β-spectrin, and protein 4.2 results in HS. The functional consequence of all these mutations is the loss of cohesion of the lipid layer with membrane skeleton leading to membrane loss.
Clinical features
HS is characterized by well-compensated mild to severe hemolytic anemia. In several instances, individuals with well-compensated anemia are diagnosed later in life as a result of the occurrence of gallstones. Although HS is a common disorder of the red cell membrane, its clinical and biologic expression at birth and in early infancy have received less attention. In a natural history study of 34 HS infants, it was shown that hemoglobin values are usually normal at birth but decrease sharply during the subsequent 20 days, which, in many cases, leads to a transient and severe anemia.5 The anemia was severe enough to warrant blood transfusions in several infants with HS (26 of 34). The aggravation of anemia seems to be related to the inability of these infants to mount an appropriate erythropoietic response to anemia and to the development of splenic filtering function, which indicates that careful monitoring of infants with HS during the first 6 months of life is important for appropriate clinical management.
Laboratory features and diagnosis
Red cell morphology is characterized by a variable number of stomatocytes in mild HS, whereas spherocytes are the dominant feature of severe HS (Figure 2). Cell dehydration of spherocytes is reflected by modest increase in mean corpuscular hemoglobin concentration (MCHC). Increased osmotic fragility as a consequence of the loss of membrane surface area is the characteristic diagnostic feature of HS. The loss of membrane surface area can be documented by an eosin-5′-maleimide binding test.3,4 Osmotic gradient deformability profiles,6 which represent measurement of cellular deformability as a continuous function of suspending medium osmolality, provide detailed insights into red cell surface area, cell surface area:volume ratio, and the state of cell hydration (Figure 3). Osmotic gradient ektacytometry is extremely useful in diagnosing HS (as shown in Figure 3) by documenting decreased cell surface area and increased osmotic fragility.6 Recently developed DNA testing panels can define the pathogenic mutations in ankyrin, band 3, α-spectrin, β-spectrin, and protein 4.2. The currently available facilities to perform DNA testing for red cell gene mutation panels are shown in Table 1.
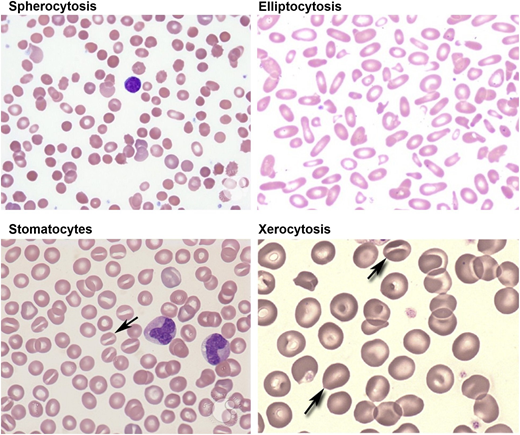
Peripheral blood smears in red cell membrane disorders. HS with marked spherocytic red cells (top left). HE with marked red cell fragmentation (top right). Hereditary OHS with stomatocytic red cells (bottom left). HX with a few target cells and variable number of stomatocytes (bottom right). Images adapted from ASH Image Bank.
Peripheral blood smears in red cell membrane disorders. HS with marked spherocytic red cells (top left). HE with marked red cell fragmentation (top right). Hereditary OHS with stomatocytic red cells (bottom left). HX with a few target cells and variable number of stomatocytes (bottom right). Images adapted from ASH Image Bank.
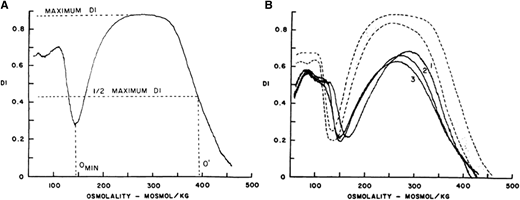
(A) Osmotic deformability profile of normal human red blood cells showing deformability index (DI) as a continuous function of suspending medium osmolality. The maximum DI, which is attained in near isotonic osmolality, is a measure of membrane surface area. Omin in the hypotonic arm corresponds to osmolality in which 50% of the red cells lyse in an osmotic fragility test, and O′ in the hypertonic arm corresponds to the state of cell hydration and hence MCHC. (B) Osmotic deformability profile of 3 patients with HS. Decreased maximum DI is the consequence of decreased membrane surface area, increase in Omin is the result of increased osmotic fragility, and decreased O′ reflects cell dehydration.
(A) Osmotic deformability profile of normal human red blood cells showing deformability index (DI) as a continuous function of suspending medium osmolality. The maximum DI, which is attained in near isotonic osmolality, is a measure of membrane surface area. Omin in the hypotonic arm corresponds to osmolality in which 50% of the red cells lyse in an osmotic fragility test, and O′ in the hypertonic arm corresponds to the state of cell hydration and hence MCHC. (B) Osmotic deformability profile of 3 patients with HS. Decreased maximum DI is the consequence of decreased membrane surface area, increase in Omin is the result of increased osmotic fragility, and decreased O′ reflects cell dehydration.
Clinical Laboratory Improvement Amendments–approved genetic diagnostic testing for erythrocyte membrane disorders in the United States, 2018
| Clinic name | Web site or contact |
|---|---|
| Mayo Clinic Medical Laboratories | https:www.mayomedicallaboratories.com |
| Molecular Genetics Laboratory, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH | kejian.zhang@cchmc.org |
| Associated Regional and University Pathologists (ARUP) Laboratories, Salt Lake City, Utah | moleculargcs@aruplb.com |
| Prevention Genetics, Marshfield, WI | clinicaldnatesting@preventiongenetics.com |
| DDC Clinic Molecular Diagnostics Laboratory, Middlefield, OH | laboratory@ddcclinic.org |
| Fulgent Diagnostics, Temple City, CA | info@fulgentgenetics.com |
| Clinic name | Web site or contact |
|---|---|
| Mayo Clinic Medical Laboratories | https:www.mayomedicallaboratories.com |
| Molecular Genetics Laboratory, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH | kejian.zhang@cchmc.org |
| Associated Regional and University Pathologists (ARUP) Laboratories, Salt Lake City, Utah | moleculargcs@aruplb.com |
| Prevention Genetics, Marshfield, WI | clinicaldnatesting@preventiongenetics.com |
| DDC Clinic Molecular Diagnostics Laboratory, Middlefield, OH | laboratory@ddcclinic.org |
| Fulgent Diagnostics, Temple City, CA | info@fulgentgenetics.com |
Treatment options
Clinical manifestations of HS can be abrogated by removal of the spleen (total splenectomy [TS]). However, because there are concerns regarding the exposure of patients to a lifelong risk of overwhelming infections and, to a lesser extent, to vascular complications after TS, the potential usefulness of subtotal splenectomy (STS) was assessed in a population of 40 patients for periods ranging from 1 to 14 years.7 After STS, a sustained decrease in hemolytic rate and continued maintenance of phagocytic function of the splenic remnant was noted. However, mild-to-moderate hemolysis was persistent and accounted for secondary gallstone formation and aplastic crisis in a subset of patients. Surprisingly, regrowth of the remnant spleen did not seem to have a major impact on the beneficial outcomes of these individuals. Because benefits of STS were different depending on the severity of the disease, a recent follow-up study showed that for children younger than age 6 years with severe anemia and/or transfusion requirement, STS was efficient and safe because it alleviated the requirement for a transfusion and increased hemoglobin to a level compatible with normal growth and activity. Of interest, half these patients did not require TS. However, the other half did require TS, but at an age when it would be much safer.
Hereditary elliptocytosis
Pathophysiology and inheritance
Hereditary elliptocytosis (HE) is another commonly inherited membrane disorder resulting from the assembly of a structurally dysfunctional red cell membrane that has decreased membrane mechanical stability.4,8 Modest weakening of lateral interactions between spectrin dimers or between elements of the spectrin-actin-protein 4.1 complex leads to elliptocytic red cells with no loss of cell surface area (nonhemolytic HE), whereas more marked weakening leads to cell fragmentation, loss of cell surface area, and decreased osmotic fragility with resultant hemolytic anemia (hemolytic HE). HE occurs in all racial groups but is particularly common in populations from West Africa.
Dominantly inherited mutations in genes encoding red cell membrane proteins, α-spectrin, β-spectrin, and protein 4.1 result in HE.8 The functional consequence of all these mutations is the weakening of the lateral interactions between spectrin dimers to form tetramers or of the interaction of protein 4.1 with spectrin and actin, which leads to decreased membrane mechanical stability.
Clinical features
HE is characterized by no anemia with only elliptocytic red cells to moderately severe hemolytic anemia with extensive cell fragmentation (Figure 2). The variable clinical expression in HE is related to the extent of decreases in membrane stability induced by skeletal protein gene mutations with heterozygous mutations that cause mild disease and double heterozygous or homozygous mutations that cause more severe disease phenotype. Interestingly, transient anemia and poikilocytosis seen during infancy seem to be related to free 2,3-bisphosphoglyceric acid (2,3-DPG) present in neonatal red cells as a consequence of diminished binding to hemoglobin F, which further decreases membrane stability and renders elliptocytic red cells more susceptible to in vivo fragmentation.9 Completion of the developmental switch from fetal to adult hemoglobin by diminishing available free 2,3-DPG can explain the abatement of poikilocytosis and hemolytic anemia by 1 year of age and manifestation as nonhemolytic HE.
Laboratory features and diagnosis
Red cell morphology is characterized by elliptocytic red cells in nonhemolytic HE, whereas cell fragmentation and poikilocytosis are features of hemolytic HE. Increased osmotic fragility is a feature of hemolytic HE. Osmotic gradient ektacytometry with characteristic deformability profiles is diagnostic for both nonhemolytic and hemolytic HE.6 Recently developed DNA testing panels can define the pathogenic mutations in α-spectrin, β-spectrin, and protein 4.1 in HE.
Treatment options
Clinical manifestations of hemolytic HE with significant anemia can be abrogated by removing the spleen. In newborn infants, it is important to ascertain whether the noted anemia with cell fragmentation is of the transient type that will evolve into nonhemolytic HE by 1 year of age.
Hereditary stomatocytosis
Hereditary xerocytosis
Pathophysiology and inheritance.
The most common red cell disorder that results from the inability to regulate normal intracellular sodium and potassium content, and hence volume homeostasis, is hereditary xerocytosis (HX).10 Passive loss of intracellular potassium in excess of a corresponding increase in sodium results in decreased total intracellular cations and obligates loss of water leading to cell dehydration. The loss of cellular deformability of HX red cells is primarily a result of increased cytoplasmic viscosity that is due to elevated intracellular hemoglobin concentration and, to a lesser extent, increased membrane rigidity, which in turn contributes to modest reduction in red cell life span.
Dominantly inherited mutations in PIEZO1, ion channel–mediating mechanosensory transduction in mammalian cells, are the most common causes of HX. In a subset of HX patients, mutations in the gene coding for calcium-activated potassium channel (KCNN4) have been identified.
Clinical features.
The HX syndrome is characterized by well-compensated mild-to-moderate hemolytic anemia. Splenomegaly is not common, although it is unclear whether it is a feature in older individuals. Some HX individuals suffer from transient perinatal edema and from pseudo-hyperkalemia. Surprisingly, iron overload in the absence of red cell transfusions is a common finding, severe enough in some individuals to require chelation. Although the mechanistic basis for iron overload is undefined, it likely reflects ineffective erythropoiesis.
Laboratory features and diagnosis.
Red cell morphology is relatively normal, with a few target cells and a variable number of stomatocytes (Figure 2). Cell dehydration is reflected by increased MCHC. Interestingly, in spite of cell dehydration, the mean cell volume is normocytic or marginally macrocytic with increased mean cell hemoglobin content. As a consequence, the red cells are osmotically resistant.11 Specialized tests can be used to document reduced red cell potassium content and total monovalent cation content. Osmotic gradient ektacytometry is diagnostic in that the profile is left shifted to lower osmolalities in the hypertonic arm of the deformability profile,6 and recently developed DNA testing panels can define the PIEZO1 and KCCN4 pathogenic mutations.
Treatment options.
At the present time, no specific treatment to correct red cell dehydration is available, and in the majority of cases, treatment is not required. However, careful monitoring of iron status seems warranted, especially in HX patients with moderate anemia and increased reticulocytes. Splenectomy is not recommended because it is not very beneficial, and it can predispose to life-threatening thrombotic episodes in some HX patients.10
OHS
Pathophysiology and inheritance.
OHS is the less common of the 2 inherited red cell membrane disorders that result from deregulated volume regulation.10 Increase of intracellular sodium in excess of corresponding decrease in potassium results in increased total intracellular cations and creates an increase in cell water content, which leads to overhydration. The loss of cellular deformability of OHS red cells is primarily the result of increased sphericity of cells, which is due to an increase in cell volume at constant membrane surface area (decreased cell surface area:volume ratio). The resultant loss of cellular deformability leads to splenic sequestration, which in turn contributes to shortened red cell life span.
Although dominantly inherited mutations in RhAG have been identified in OHS, the genetic and mechanistic basis for OHS in contrast to HX has yet to be fully defined.
Clinical features.
The OHS syndrome is characterized by mild-to-moderate hemolytic anemia with reticulocytosis. Because far fewer cases of OHS have been reported compared with HX, the clinical features other than hemolytic anemia are less well defined.
Laboratory features and diagnosis.
Stomatocytes are a major feature of red cell morphology (Figure 2). Cell hydration is reflected by increased mean cell volume and decreased MCHC. As a consequence, the red cells are osmotically fragile. Specialized tests can be used to document increased red cell sodium content and total monovalent cation content. Osmotic gradient ektacytometry is diagnostic in that the profile is right shifted to higher osmolalities in the hypertonic arm of the deformability profile.6
Treatment options.
At the present time, there are no specific treatments to correct increased red cell hydration. Because increased iron overload has been implicated in some cases of OHS, a careful monitoring of iron status seems warranted. Splenectomy is not recommended because it is not very beneficial and, in addition, it predisposed some OHS patients to life-threatening thrombotic episodes.12
Final considerations
Detailed molecular and mechanistic insights into the pathophysiology of inherited red cell membranes have been developed (Table 2). As a result, significant advances have been made in diagnosis and clinical management of inherited red cell membrane disorders that result in hemolytic anemia.
Red cell membrane disorders
| Disorder | Severity | Inheritance | Molecular defects | Morphology | Osmotic fragility | Splenectomy |
|---|---|---|---|---|---|---|
| HS | Mild to severe | AD, AR, de novo | Ankyrin-1, band 3, α-spectrin, β-spectrin, protein 4.2 | Varying degree of spherocytes | Mild to marked decrease | Beneficial |
| HE | Nonhemolytic to severe | AD | α-spectrin, β-spectrin, protein 4.1 | Elliptocytes and fragmented red cells | Normal to marked decrease | Beneficial |
| OHS | Mild to moderate | AD | RhAG | Stomatocytosis | Increased | Not recommended |
| HX | Nonhemolytic to moderate | AD | Piezo-1, Gardos channnel | Some target cells | Decreased | Not recommended |
| Disorder | Severity | Inheritance | Molecular defects | Morphology | Osmotic fragility | Splenectomy |
|---|---|---|---|---|---|---|
| HS | Mild to severe | AD, AR, de novo | Ankyrin-1, band 3, α-spectrin, β-spectrin, protein 4.2 | Varying degree of spherocytes | Mild to marked decrease | Beneficial |
| HE | Nonhemolytic to severe | AD | α-spectrin, β-spectrin, protein 4.1 | Elliptocytes and fragmented red cells | Normal to marked decrease | Beneficial |
| OHS | Mild to moderate | AD | RhAG | Stomatocytosis | Increased | Not recommended |
| HX | Nonhemolytic to moderate | AD | Piezo-1, Gardos channnel | Some target cells | Decreased | Not recommended |
AD, autosomal dominant; AR, autosomal recessive.
Correspondence
Narla Mohandas, New York Blood Center, 310 E 67th St, New York, NY 10065; e-mail: mnarla@nybc.org.
