
Abstract
The myelodysplastic syndromes are collectively the most common myeloid neoplasms. Clonal hematopoiesis present in these diseases results in bone marrow failure characteristically seen in patients. The heterogeneity of myelodysplastic syndrome pathobiology has historically posed a challenge to the development of newer therapies. Recent advances in molecular characterization of myelodysplastic syndromes are improving diagnostic accuracy, providing insights into pathogenesis, and refining therapeutic options for patients. With the advent of these developments, appropriately chosen therapeutics or even targeted agents may be able to improve patient outcomes in the future.
Learning Objectives
Describe the current understanding of somatic mutations in MDS and the implications for disease biology that can be used therapeutically
Discuss the current status of novel agents targeted against molecular features for the treatment of MDS
Explain the molecular rationale for guiding use of currently FDA-approved medications for MDS
Introduction
The myelodysplastic syndromes (MDS) are a heterogeneous collection of clonal hematopoietic malignancies that comprise a large subgroup of the myeloid neoplasms and collectively are the most common acquired adult bone marrow failure syndromes.1 They are characterized by poor overall survival (OS) due to ineffective hematopoiesis, progressive cytopenias, and transformation to acute myeloid leukemia (AML). Current therapeutics for MDS are primarily based on stratification into lower- and higher-risk disease using clinical prognostic scoring systems. Currently, only 3 agents are approved for the treatment of MDS by the US Food and Drug Administration (FDA): azacitidine (AZA), decitabine (DAC), and lenalidomide (LEN) and none based on molecular features of the disease. Allogeneic bone marrow transplantation (BMT) remains the only cure. Over the past 10 years, high-throughput DNA sequencing of somatic mutations has advanced our understanding of MDS biology, and a paradigm shift in the management of MDS may be impending. Our increasing knowledge of the molecular biology of MDS may predict response to extant therapies and lead to a more rational approach to treatment. Even more exciting is the potential for novel therapies based on our newfound molecular knowledge. This review will describe the current knowledge of somatic mutation patterns in MDS and the implications for potential targeted treatment strategies and guidance with our current therapeutic tools.
The biology of MDS
Cytogenetics have been examined in myeloid malignancies since the 1970s. Karyotypic abnormalities have long been incorporated into prognostic scoring systems2 and, more recently, certain cytogenetic alterations have guided therapies, such as lenalidomide in del5q MDS.3 Genetic technology has advanced rapidly, and next-generation DNA sequencing is routinely available for clinical application.4 The information available from next-generation DNA sequencing has exciting implications for the treatment of MDS.
The overall pattern of molecular mutations observed in MDS is similar to what has been described in other diseases. A few genes are mutated with relatively higher frequencies whereas many other genes are mutated only in a small minority of patients (Figure 1). Consequentially, genes with potential broad clinical applicability are those that are more frequent (occurring in a significant percentage of patients) or those with markedly strong or consistent effects when present. In contrast to some diseases, no single gene mutation accounts for the majority of MDS cases; most genes are mutated in <5% of patients.5,6 It has become apparent that genetically MDS overall are mostly diseases of epigenetic dysregulation and disordered spliceosome function. Other frequently implicated pathways include DNA damage response and repair enzymes, hematopoietic transcription regulation, cell-signaling pathways, and cohesin genes. Adding complexity is the observation that clinical MDS phenotypes are much more closely tied to the individual genes mutated that the pathways into which these genes fall.7 In other myeloid diseases such as AML or myeloproliferative neoplasms (MPNs), somatic mutation testing is increasingly considered to be standard of care at diagnosis, and perhaps throughout the disease course, allowing access to therapies targeted at underlying disease biology. Midostaurin in FLT3-mutated AML is one of the more recent examples achieving US regulatory approval in April 2017.8 Unfortunately, establishing similar therapeutic schema using molecular mutations for drug targeting has been less straightforward in MDS due in large part to the clinical and genetic heterogeneity of MDS. A particular challenge in MDS is that only a handful of mutations in this disease are gain-of-function mutations and thus amenable to targeted inhibition. We are therapeutically better at inhibiting proteins but less adept at improving their performance; thus, loss-of-function mutated diseases cannot be easily targeted and these are the majority of mutations in MDS. Nonetheless, somatic mutations of individual genes will increasingly play critical roles in MDS disease-specific assessments and treatment paradigms.

Recurrent somatic mutations in MDS by pathway. Includes approximate frequency of the most common recurrent somatic mutations in MDS and their prognostic significance. Some mutations influence the phenotype and are therefore more common in specific subtypes of MDS; for instance, SF3B1 mutations are found in up to 80% of patients with MDS-RS and SRSF2 mutations are more common in MPN/MDS overlap syndromes such as chronic myelomonocytic leukemia. Mutation frequencies and their prognostic significance are adapted from Bejar et al,32 Haferlach et al,5 and Papaemmanuil et al6 published frequencies. The negative prognostic impact with SRSF2 mutations is adapted from Thol et al.63
Recurrent somatic mutations in MDS by pathway. Includes approximate frequency of the most common recurrent somatic mutations in MDS and their prognostic significance. Some mutations influence the phenotype and are therefore more common in specific subtypes of MDS; for instance, SF3B1 mutations are found in up to 80% of patients with MDS-RS and SRSF2 mutations are more common in MPN/MDS overlap syndromes such as chronic myelomonocytic leukemia. Mutation frequencies and their prognostic significance are adapted from Bejar et al,32 Haferlach et al,5 and Papaemmanuil et al6 published frequencies. The negative prognostic impact with SRSF2 mutations is adapted from Thol et al.63
Using somatic mutations to consider targeted therapy
There are many potential pathways to target in the pathobiology of MDS, including cell signaling pathways, cell cycle regulation pathways, inflammatory pathways,9 and innate immune pathways among others.10 Increasingly, these pathways (and the mutations affecting them) are considered to be potential therapeutic targets. Figure 2 shows select current and future potential pharmaceutical targets in MDS. It is important to emphasize that none of these drugs have completed phase 3 MDS studies; most are in earlier phase studies in patients with refractory or relapsed disease. Luspatercept is the furthest along with on ongoing phase 3 trial. Nonetheless, the fact that early-phase studies are proceeding is encouraging, especially as no drugs have been approved for higher-risk MDS with progression during hypomethylating agent (HMA) therapy or an insufficient response to HMA therapy. It should be a sobering reminder that patients in whom standard HMA therapy has failed, median survival is 5.6 months, with a 2-year survival probability of 15%.11,12 This includes primary and secondary HMA failures as well as patients in whom the treatment is intolerable. As we gain experience with these novel agents and their toxicities, targeted therapies may be potentially deployed earlier in therapeutic pathways and hopefully modify the natural history of MDS. The development of targeted therapies is not a fool’s errand but a noble quest. Table 1 shows some currently available clinical trials in this arena.
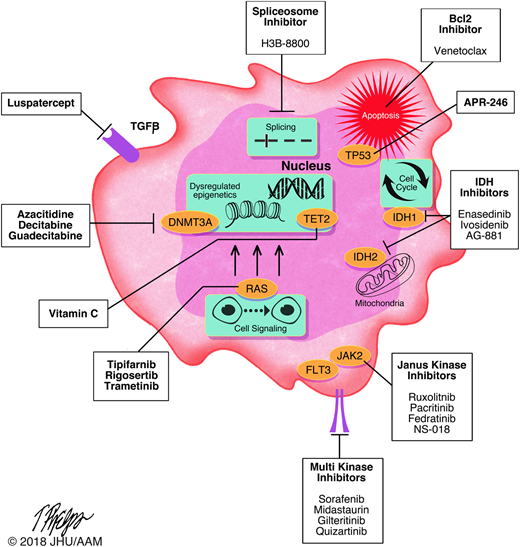
Molecular targeted therapies in MDS. Includes select investigational and approved therapies for patients with MDS and their respective targets.
Molecular targeted therapies in MDS. Includes select investigational and approved therapies for patients with MDS and their respective targets.
Select therapeutic studies that allow MDS patient enrollment with eligibility specifying molecular features
| Mutation targeted | Drug | Mechanism of action | Patient type | Phase and trial name | NCT identifier |
|---|---|---|---|---|---|
| SF3B, SRSF2, U2AF1, or ZRSR2 | H3B-8800 (oral) | Inhibitor of the splicing factor SF3B1 | AML: not candidates for induction | Phase 1 Trial to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of Splicing Modulator H3B-8800 for Subjects with MDS, AML, and CMML | NCT02841540 |
| MDS: HMA refractory if higher risk or transfusion dependent if lower risk | |||||
| CMML: therapy refractory | |||||
| TET2 | Vitamin C (IV) | Mimics TET2 restoration by promoting | MDS patients with <20% blasts and platelets >20 000 (allows concurrent HMA) | Phase 1b/2a Tolerability of Vitamin C in Patients with Intermediate or High Risk MDS with TET2 Mutations | NCT03433781 |
| DNA demethylation and reversing aberrant stem cell self renewal. | |||||
| TET2 | Ascorbic acid (oral) | As above (in combination with AZA) | MDS, MDS/MPN, or AML patients who are HMA naive | Phase 2 TET2 Mutations in Myelodysplastic Syndromes and Acute Myeloid Leukemia with Azacitidine + Ascorbic Acid | NCT03397173 |
| TP53 | APR-246 (IV) | Reactivate mutant p53 to induce programmed cell death | Treatment-naive myeloid neoplasm patients with <30% blasts | Phase 1b/2 Safety and Efficacy of APR-246 w/Azacitidine for tx of TP53 Mutant Myeloid Neoplasms | NCT03072043 |
| IDH2 | Enasidenib (oral) | Reduces the oncometabolite, 2-HG through inhibition of mIDH2 protein | MDS patients (up to 30% blasts) naive to HMA (arm A) or R/R to 6 cycles of HMA | Phase 2 Targeted Therapy with the IDH2-Inhibitor Enasidenib (AG221) for High-Risk IDH2-Mutant Myelodysplastic Syndrome | NCT03383575 |
| IDH1 | FT-2102 (oral) | Undefined mechanism | AML or MDS (INT, HIGH, VERY HIGH by IPSS-R) disease R/R to previous therapy | A Phase 1/2, Multicenter, Open-label Study of FT-2102 as a Single Agent and in Combination with Azacitidine or Cytarabine in Patients with Acute Myeloid Leukemia or Myelodysplastic Syndrome with an IDH1 Mutation | NCT02719574 |
| IDH1 R132 | Ivosidenib (oral) | Reduces the oncometabolite, 2-HG through inhibition of mIDH1 protein | R/R advanced hematologic malignancies patients | Phase 1 Study of Orally Administered AG-120 in Subjects with Advanced Hematologic Malignancies with an IDH1 Mutation | NCT02074839 |
| IDH2 | Enasidenib (oral) | Reduces the oncometabolite, 2-HG through inhibition of mIDH2 protein | R/R advanced hematologic malignancies patients (untreated arm if patients unfit) | Phase 1/2 Study of AG-221 in Subjects with Advanced Hematologic Malignancies with an IDH2 Mutation | NCT01915498 |
| IDH1 or IDH2 | AG-881 (oral) | Small-molecule mIDH1 and mIDH2 protein inhibitor; reduces the oncometabolite, 2-HG | R/R advanced hematologic malignancies patients | Study of Orally Administered AG-881 in Patients with Advanced Hematologic Malignancies with an IDH1 and/or IDH2 Mutation | NCT02492737 |
| IDH1 R132 | Venetoclax + Ivosidenib (oral) | BCL2 inhibition in combination with inhibition of mIDH1 protein | R/R AML; Patients with high-risk MDS or MPN (defined as ≥10% bone marrow blasts) may also be eligible after discussion with investigator | Study of Venetoclax with the mIDH1 Inhibitor Ivosidenib (AG120) in IDH1-Mutated Hematologic Malignancies | NCT03471260 |
| Mutation targeted | Drug | Mechanism of action | Patient type | Phase and trial name | NCT identifier |
|---|---|---|---|---|---|
| SF3B, SRSF2, U2AF1, or ZRSR2 | H3B-8800 (oral) | Inhibitor of the splicing factor SF3B1 | AML: not candidates for induction | Phase 1 Trial to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of Splicing Modulator H3B-8800 for Subjects with MDS, AML, and CMML | NCT02841540 |
| MDS: HMA refractory if higher risk or transfusion dependent if lower risk | |||||
| CMML: therapy refractory | |||||
| TET2 | Vitamin C (IV) | Mimics TET2 restoration by promoting | MDS patients with <20% blasts and platelets >20 000 (allows concurrent HMA) | Phase 1b/2a Tolerability of Vitamin C in Patients with Intermediate or High Risk MDS with TET2 Mutations | NCT03433781 |
| DNA demethylation and reversing aberrant stem cell self renewal. | |||||
| TET2 | Ascorbic acid (oral) | As above (in combination with AZA) | MDS, MDS/MPN, or AML patients who are HMA naive | Phase 2 TET2 Mutations in Myelodysplastic Syndromes and Acute Myeloid Leukemia with Azacitidine + Ascorbic Acid | NCT03397173 |
| TP53 | APR-246 (IV) | Reactivate mutant p53 to induce programmed cell death | Treatment-naive myeloid neoplasm patients with <30% blasts | Phase 1b/2 Safety and Efficacy of APR-246 w/Azacitidine for tx of TP53 Mutant Myeloid Neoplasms | NCT03072043 |
| IDH2 | Enasidenib (oral) | Reduces the oncometabolite, 2-HG through inhibition of mIDH2 protein | MDS patients (up to 30% blasts) naive to HMA (arm A) or R/R to 6 cycles of HMA | Phase 2 Targeted Therapy with the IDH2-Inhibitor Enasidenib (AG221) for High-Risk IDH2-Mutant Myelodysplastic Syndrome | NCT03383575 |
| IDH1 | FT-2102 (oral) | Undefined mechanism | AML or MDS (INT, HIGH, VERY HIGH by IPSS-R) disease R/R to previous therapy | A Phase 1/2, Multicenter, Open-label Study of FT-2102 as a Single Agent and in Combination with Azacitidine or Cytarabine in Patients with Acute Myeloid Leukemia or Myelodysplastic Syndrome with an IDH1 Mutation | NCT02719574 |
| IDH1 R132 | Ivosidenib (oral) | Reduces the oncometabolite, 2-HG through inhibition of mIDH1 protein | R/R advanced hematologic malignancies patients | Phase 1 Study of Orally Administered AG-120 in Subjects with Advanced Hematologic Malignancies with an IDH1 Mutation | NCT02074839 |
| IDH2 | Enasidenib (oral) | Reduces the oncometabolite, 2-HG through inhibition of mIDH2 protein | R/R advanced hematologic malignancies patients (untreated arm if patients unfit) | Phase 1/2 Study of AG-221 in Subjects with Advanced Hematologic Malignancies with an IDH2 Mutation | NCT01915498 |
| IDH1 or IDH2 | AG-881 (oral) | Small-molecule mIDH1 and mIDH2 protein inhibitor; reduces the oncometabolite, 2-HG | R/R advanced hematologic malignancies patients | Study of Orally Administered AG-881 in Patients with Advanced Hematologic Malignancies with an IDH1 and/or IDH2 Mutation | NCT02492737 |
| IDH1 R132 | Venetoclax + Ivosidenib (oral) | BCL2 inhibition in combination with inhibition of mIDH1 protein | R/R AML; Patients with high-risk MDS or MPN (defined as ≥10% bone marrow blasts) may also be eligible after discussion with investigator | Study of Venetoclax with the mIDH1 Inhibitor Ivosidenib (AG120) in IDH1-Mutated Hematologic Malignancies | NCT03471260 |
Current as of 30 April 2018 on clinicaltrials.gov, these trials are listed as “active” or “pending.”
2-HG, 2-hydroxyglutarate; R/R, relapsed and/or refractory.
Pre-mRNA splicing pathways and potential targeting
RNA splicing mutations are present in up to 45% of MDS.13 These mutations most often affect the factors that recognize 3′ consensus splice sites. Mutations in the SF3B1 gene are common (∼20% of MDS) and are associated with ring sideroblasts on morphological examination of the marrow, lower-grade disease, and better prognosis.14 Patients with MDS with ring sideroblasts without mutated SF3B1 (∼20% of patients) are thought to have an inferior prognosis compared with patients with mutated SF3B1.15 Other splicing mutations exist as well but less is known about U2AF1, SRSF2, and ZRSR2. Mutations in the SRSF2 gene are present in ∼10% of patients with MDS and are associated with a poor prognosis but are more prevalent in chronic myelomonocytic leukemia (CMML) at 40% to 50% of cases.16 Mutations in U2AF1 are present in 8% to 12% of MDS and are believed to convey a less favorable prognosis. U2AF1 mutations have been shown to alter splice recognition sites and specificity of precursor messenger RNA (mRNA) binding, eliciting changes in thousands of RNA transcripts; however, the exact mechanism by which U2AF1 mutations give rise to dysplasia is yet to be determined.17 Lastly, mutations in ZRSR2 are present in 5% of patients with MDS and also demonstrate a poor prognosis. Mutations affecting these different splicing factors lead to distinct aberrant splicing, although no common misspliced isoform has been found to be pathogenic; however, the misspliced protein does not need to pathogenic to be targetable. The mutated cells may be addicted to certain isoforms that are not pathogenic themselves but could be targetable. Treatment of human AML xenografts with mutant SRSF2 in nonobese diabetic (NOD) SCID γ mice with the spliceosome modifying compound E7107 demonstrated significant reduction in leukemia burden compared with wild-type xenografts. A phase 1/2 study is currently under way to evaluate the splicing inhibitor H3B-8800 in patients with splicing gene–mutant MDS and AML (NCT02841540) (Table 1).
In addition to allowing the development of agents that specifically target the “product” of mutated genes, an increased understanding of the broader role of mutations to MDS pathophysiology may allow optimum use of other therapeutic strategies. For example, it has recently been shown that the transforming growth factor-β (TGF-β) superfamily are potent regulators of erythropoiesis with a role in the ineffective erythropoiesis of MDS. TGF-β ligands trigger receptor-mediated phosphorylation and activation of the inhibitory Smad2/3 transcription factors, leading to suppression of terminal erythroid differentiation. Luspatercept is a novel fusion protein that blocks TGF-β superfamily inhibitors of erythropoiesis as it is a recombinant human counterpart that contains the modified extracellular domain of the activin receptor IIB. The luspatercept PACE-MDS trial was a phase 1/2 multicenter, open-label, dose-finding study of 58 patients with lower-risk (by International Prognostic Scoring System18 ) MDS (27 in dose escalation and 31 in expansion phase). In the dose-escalation phase, transfusion independence was achieved in 35% of patients receiving higher doses of treatment (0.75-1.75 mg/kg subcutaneously every 21 days). A key finding was the higher erythroid response rate in patients with ring sideroblasts (55% vs 29% in ring sideroblasts–negative) and 60% of those with a SF3B1 gene mutation.19 This has led to a phase 3, randomized, double-blind study comparing luspatercept to placebo in transfusion-dependent, low/intermediate-risk patients with MDS with ring sideroblasts, referred to as the MEDALIST trial (NCT02631070). Notably, this is not a targeted mechanism of action; rather, this trial has provided guidance toward a more effective agent in a molecular (SF3B1) and morphologic (sideroblasts) subset of patients. Additionally, luspatercept has clinical utility in several other MDS subtypes as well. A similar finding, albeit at the karyotypic level, is the 27% response rate to lenalidomide in non-delq5 patients.20 These examples are a cautionary tale against not restricting drug indications too specifically according to mutations, as there may be beneficial applications in patients without the mutation as well, given that we still have a relatively limited understanding of the role of some of these mutations.
Epigenetic regulation pathways and potential targeting
Mutations involving DNA methylation including those in TET2, DNMT3A, and IDH1/2 are common in MDS and other myeloid conditions. In general, these TET2 and IDH mutations are mutually exclusive within a given patient. The TET2 gene encodes a protein involved in the conversion of 5-methyl-cytosine to 5-hydroxymethyl-cytosine, which is pivotal for DNA demethylation.21 TET2 mutations are generally associated with a normal karyotype and are of unclear prognostic significance.22 TET2 is currently thought to be the most commonly mutated gene in MDS. This makes it not only potentially attractive for targeting but also a target we should approach carefully. Interestingly, TET2 is thought to be “targetable” if not with a novel drug, but with vitamin C.23 Recently, it has been shown that treatment with vitamin C mimics TET2 function and restores hematopoiesis in mouse and human cells with TET2 deficiency.23,24 However, this finding had never been tested in vivo or in the context of hematopoietic cells. This effect has just started to be explored in early-phase trials (Table 1) alone or in combination with HMAs (NCT03433781; NCT03397173). Caution should be applied clinically, however, outside of vitamin C’s dosing preset by well-monitored clinical trials as high doses of vitamin C increase the risk calcium oxalate nephrolithiasis in humans.
DNMT3A encodes a protein that catalyzes the transfer of methyl groups required for de novo methylation. Mutations in DNMT3A are slightly less common than TET2, and are estimated to occur in 12% to 18% of MDS cases.25 DNMT3A mutations are thought to be an early event in clonal hematopoiesis. Their exact mechanism and function is unclear but appear to act in a dominant-negative manner.26 Most DNMT3A mutations involve the p.R882 codon.27 Mutations in DNMT3A may be associated with a poorer prognosis in MDS but this is inconsistent as paradoxically, DNMT3A mutations may remain during complete remission without adverse outcome.28 The early action of DNMT3A in clonal hematopoiesis may prevent it from being a therapeutic target, as the consequence of early interference in clonal hematopoiesis could possibly have significant consequences. This could be related to the lack of normal hematopoietic progenitors with a germ line configuration. One could also hypothesize that once transformed, the cells do not depend on mutated DNMT3A anymore but this mutation does predispose to additional hits but may not be oncogenic themselves.
Mutations in the isocitrate dehydrogenases IDH1 and IDH2 are generally more common in AML than in MDS, where they are present in <5% of cases. Isocitrate dehydrogenases catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate; mutations in the catalytic domains of IDH1/2 result in the accumulation of 2-hydroxyglutarate, resulting in DNA hypermethylation.29 Mutant IDH enzymes have neomorphic activity and catalyze reduction of α-KG to the (R) enantiomer of 2-hydroxyglutarate, which is associated with DNA and histone hypermethylation, altered gene expression, and blocked differentiation of hematopoietic progenitor cells. The prognostic significance of mutant IDH (mIDH) is controversial but appears to be influenced by comutational status and the specific location of the mutation (IDH1-R132, IDH2-R140, IDH2-R172). Importantly, these have been an area of active investigation for targeting and have shown the most promise in this arena. Enasidenib (AG-221), a selective small molecule mutant IDH2 inhibitor, was recently approved for relapsed or refractory IDH2-mutated AML (not MDS) based on the results from a phase 1/2 study of 199 patients. In that study, 19.3% of patients achieved a complete response (CR), with a median CR duration of 8.2 months.30 Regarding its activity in MDS, enasidenib was evaluated in 17 patients with relapsed or refractory MDS in a phase 1 study, which reported an ORR of 53%, including 1 CR. Of the 10 patients who had previously received HMA therapy, 5 (50%) experienced clinical benefit with enasidenib and 1 had a CR. There is now an ongoing trial of enasidenib specifically in higher-risk MDS patients. (NCT03383575). There are also trials of agents for high-risk mIDH1 patients (NCT02719574, NCT02074839) as well as a small molecular inhibitor of both mIDH1 and mIDH2, AG-881(NCT02492737). The role of ivosidenib for IDH1-mutated AML to induce durable remissions has been recently demonstrated.31 In patients who received an oral daily dose of 500 mg ivosidenib in the primary efficacy population (179 patients), the rate of complete remission or complete remission with partial hematologic recovery was 30.4% (95% confidence interval [CI], 22.5-39.3), the rate of complete remission was 21.6% (95% CI, 14.7-29.8), and the overall response rate was 41.6% (95% CI, 32.9-50.8). Grade 3 adverse events were seen in nearly 21% of this population. Most notable adverse events were differentiation syndrome and Qt interval prolongation. There were 12 MDS patients included in this treatment dose and were noted to be recurrent or refractory after HMA. Five of these 12 were noted to achieve CR, showing promise for this drug in MDS specifically. There is also interest in combination therapy to achieve dual mechanism of actions between targets as in a new trial that will combine venetoclax (a bcl2 inhibitor) and ivosidenib in myeloid malignancies (NCT03471260).
Transcription factor pathways and potential targeting
Commonly mutated transcriptional regulation genes in MDS include RUNX1 and TP53 whereas MECOM and GATA2 mutations are less common. RUNX1 is a transcription factor that regulates myeloid development and is mutated in ∼10% of patients with MDS. Mutations including copy losses in the RUNX1 gene are generally associated with a poor prognosis.32 Germ line RUNX1 deficiency is also associated with thrombocytopenia from an inherited condition known as familial platelet disorder with predisposition to myeloid leukemias.33 Thus far, targeting in this arena has not been investigated.
Mutations in TP53 have been reported in 5% to 18% of patients with MDS and are generally associated with higher-risk disease, including MDS with excess blasts and therapy-related myeloid neoplasms as well as complex cytogenetics and a small subset of patients with 5q minus syndrome.34 TP53 mutations are considered a universally poor prognostic factor. In targeting mutant p53, a group of compounds known as p53 activators are promising therapeutic agents whose mechanism of action is to restore the wild-type conformation of mutant p53 and thereby rescue of p53 function.35 APR-246 is a p53 reactivator that has been investigated in a previous phase 1 study with activity in TP53-mutant AML.36 There is now an accruing phase 1b/2 study of APR-246 in combination with azacitidine (NCT03072043) for MDS (or myeloid neoplasm) patients with up to 30% blasts. Given the poor outcomes in TP53 patient groups, this is an area for optimism if the trial yields favorable results.
Cell signaling and signal transduction
Signal transduction genes as a group are less commonly mutated in MDS than in other myeloid neoplasms such as AML or MPNs. The JAK2 gene is a nonreceptor tyrosine kinase that acts through the STAT-signaling pathway; the most common variant is a single mutational hotspot, p. V617F. Though common in polycythemia vera and essential thrombocytosis, JAK2 mutations are present in fewer than 5% of patients with MDS and are enriched in cases of myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis.37 Conversely JAK2 V617F mutations are present in ∼90% to 95% of patients with polycythemia vera as well as more than 50% of patients with essential thrombocytosis and idiopathic myelofibrosis. The overall prognostic significance of JAK2 mutations is uncertain in MDS but it is not uncommon to think about a role for targeting here given what we have learned in MPNs. Other signal transduction genes such as Ras family members including NRAS, and less commonly KRAS, are mutated in 5% to 10% of patients with MDS and are more frequent in patients with CMML.38 Both NRAS and KRAS mutations occur at known hotspots, including p.G12, p.G13, and p.Q61. Mutations in NRAS likely confer a less favorable prognosis in MDS. It is known that when acquired at times of clonal evolution mutations in these signal transduction genes in MDS patients are associated with increased progression to acute leukemia.39
Given this aggressive nature, targeting these mutations has appeal through direct mutational applications or other involved pathways including PI3K or MEK. Both JAK2 and RAS mutations have been targeted with some success in non-MDS and other myeloid diseases, leading to interest in potential application in MDS as well. Trials investigating these pathways include tipifarnib in CMML (NCT02807272), rigosertib in MDS,40 and trametinib in refractory myeloid diseases41 Ruxolitinib, pacritinib, fedratinib, and NS-018 have been best studied in MPNs. Although quite common in AML, FLT3 mutations are rare events in MDS. Despite the recent approval for midostaurin in this AML setting and multiple other agents studied for FLT3-mutated AML (sorafenib, gilteritinib, quizartinib), this target is less likely to play a role in MDS therapeutics given the lower prevalence.
Histone modifiers
Lastly, mutations in EZH2 have been reported in 5% to 10% of patients with MDS42 and are associated with a poor prognosis. An EZH2 inhibitor tazemetostat has ongoing studies in lymphoma and other tumors whereas DS-3201, EZH1/2 inhibitor is being explored in AML. It is more likely the latter inhibitor would have relevance in MDS as the myeloid EZH2 mutations are distinct from the lymphoid mutations with loss of function compared with gain-of-function roles. Future investigations for therapeutic potential in MDS are warranted. ASXL1 is mutated in 15% to 25% of patients with MDS and is also commonly mutated in AML. Mutations in the ASXL1 gene are associated with a poor prognosis and have also been implicated in rare cases of familial MDS.43 Mutations in histone-modifier genes are common in MDS and appear to be enriched in CMML. Given the often higher-risk nature of MDS patients with ASXL1, investigations agents with this target in mind are welcome. Recent investigations in a transgenic mouse model have implicated a gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies.44 This suggests a biologic rationale for the use of bromodomain inhibitors in ASLX1-mutated MDS.
Using somatic mutations to guide standard therapy
With the recent excitement about the above novel agents but in the absence of approved agents targeting somatic mutations, there is also interest in using molecular testing as guidance for currently available therapies. This knowledge has been gleaned from retrospective analyses of datasets of nontargeted standard therapies, looking for associations between response to therapy and somatic mutations.
Lenalidomide
The first genetic abnormality guiding management decisions in MDS was the interstitial deletion involving the long arm of chromosome 5 (del5q MDS). Lenalidomide selectively suppresses del(5q) clones in MDS through induction of ubiquitination of casein kinase 1A1 (CK1α) encoded within a commonly deleted region resulting in CK1α degradation and subsequent erythroid growth arrest.45,46 The “targeted” use of lenalidomide del5q MDS yielded a 50% or greater reduction in transfusions in 76% of patients, whereas 67% achieved transfusion independence lasting a median duration of >2.7 years.47 This type of response provides an encouraging template for future “targeted” therapeutic efforts. Ironically though, the decision to use this therapy as well as the response rates came before the biology of the therapeutic effect was known. Also, it should be remembered that there is a 27% response rate to lenalidomide in non delq5 patients, emphasizing the limits of predicting efficacy of targeted therapy.20 Further guidance in del5q MDS also comes from NGS; patients with del5q as well as TP53 mutations do less well compared with del5q patients without TP53.34,48
Response to hypomethylating agents
Although azacitidine treatment improves overall survival compared with conventional care, up to 50% of patients will not respond to treatment with HMAs. In a retrospective study, 213 patients with MDS receiving treatment with HMAs had mutational profiling.49 In this cohort, 94% of patients carried at least 1 mutation, with ASXL1 (46%) the most frequent, followed by TET2 (27%), RUNX1 (20%), TP53 (18%), and DNMT3A (16%). Though there was a trend favoring a higher response rate in patients with TET2 mutations, it was not significant until the analysis was limited to those patients with allele frequencies >10%. At the higher variant allele frequency, TET2 mutations were associated with a significantly higher response rate (60%) compared with wild type (43%; P = .036). Furthermore, the presence of mutated TET2 and wild-type ASXL1 had the highest response rate, whereas those patients with mutated ASXL1 and wild-type TET2 had a trend toward a lower response rate (P = .051). A complex karyotype with a TP53 mutation was associated with poor overall survival, whereas complex karyotypes with wild-type TP53 had the same survival as other karyotypes. Interestingly, mutations involving RUNX1, ASXL1, EXH2, and ETV6 did not significantly influence prognosis, suggesting that treatment with HMAs may modify the unfavorable impact of these mutations. Although this study identified somatic mutations that that may impact HMA response, it did not result in guidance for mutations which could predict primary resistance and thus suggesting paths of therapy away from HMAs. Another retrospective study of 134 patients with higher-risk MDS treated with AZA showed an association between karyotype and mutation profile with overall survival.50 Mutations involving histone modifiers, including ASXL1, EZH2, and MLL, were positively associated with prolonged survival (P = .001). Specifically, patients with mutations in histone modifiers without high-risk cytogenetics had a response rate of 79% and median survival of 29 months compared with a response rate of 49% and 10-month median survival in the same patients with high-risk cytogenetics (P < .001). TP53 mutations were again a significant unfavorable covariate for overall survival (P = .001).50
Two recent studies suggest that TP53-mutant MDS/AML might be best treated with decitabine. In a study of 116 patients with MDS/AML, outcomes were explore with a 10-day course of decitabine every 28 days.51 Patients with a TP53 mutations had a significantly higher overall response rate compared with wild type (21 [100%] of 21 patients vs 32 [41%] of 78 patients; P < .001) and higher rate of complete remission/incomplete marrow recovery (CR/CRi; 13 [62%] of 21 patients vs 26 [33%] of 78 patients; P = .04). In another retrospective study evaluating 109 patients with MDS treated with decitabine, TP53 mutations were identified in 13.8% of patients.52 TP53 was the only somatic gene mutation predictive for CR, with 10 of 15 patients with TP53 mutations (66.7%) achieving CR vs 20 (21%) of 94 with wild type (P = .001). Of those with monosomies, 80% achieved CR. Median OS remained disappointing at 14 months. These favorable response rates have not been uniformly seen in TP53-mutant disease treated with decitabine in all studies so these result may be challenging to duplicate in a real-world patient. Additionally, TP53 mutant clones probably do not display exclusive sensitivity to decitabine compared with azacitidine so use of either HMA remains reasonable but clearly better therapies for TP53 mutated disease are warranted.
Allogeneic BMT
BMT remains the only curative treatment strategy for patients with MDS. Recent studies have found that specific MDS-related mutations are associated with poor survival after BMT, regardless of patient specific factors such as age, performance status, and even disease staging.53 Certain somatic mutations seem to influence the probability of relapse after BMT.53 In a study of 401 patients with MDS or secondary AML who underwent allogeneic BMT, the number and type of somatic mutations significantly affected outcome.54 Mutations involving RUNX1, ASXL1, or TP53 were independent markers of relapse after transplantation. Patients with TP53 mutations had a particularly poor outcome. In a Bone Marrow Transplant Clinical Trials Network US study of 1514 patients with MDS who had undergone BMT from 2005 to 2014, TP53 mutations were again found to be the most powerful predictors of poor posttransplant survival.55 Myeloablative conditioning did not result in a lower rate of relapse or death in these patients compared with that of patients who had received reduced intensity conditioning. A study of 617 Japanese patients with MDS and secondary AML also found unfavorable outcomes post BMT for patients with TP53 mutations, in addition to RAS-pathway mutations.56 Lastly, a recent study of over 300 German patients found mutated TP53 and/or complex karyotype NRAS, IDH1, and EZH2 to have a negative prognostic effect on posttransplant relapse.57
Potential limitations to targeted therapies in MDS
Polyclonality at diagnosis and clonal evolution through the disease timeline may be the biologic issues that limit targeting single mutations in MDS. Sequencing-based studies have demonstrated that MDS is composed of a founding clone. Mutations can be assigned to clusters and can be used to determine the clonal composition of the given MDS disease in a patient. Variant allele frequencies (VAFs) often reported for mutational burden in MDS play a role in this, but we have an incomplete understanding at present. Reconstruction of MDS clonal architecture requires nonbiased sequencing approaches such as exome or whole-genome sequencing of tumor and normal germline tissue to have a sufficient number of mutations for accurate clone assignment. This is not currently clinically standard nor is it likely to have bedside relevance any time soon. The relationship between tumor clonality and disease progression is not well understood at present, though the risk of MDS progression to AML has been associated with more subclones in at least 1 study.58 Serial sequencing of MDS bone marrow samples allows for more accurate assessment of tumor clonality by comparing the patterns of VAF changes over time, primarily in a research context. Serial bone marrow sequencing studies have also demonstrated that somatic mutations can be detected at low levels when MDS patients are in complete remission but ongoing detection of mutations, at least in AML, after therapy has been predictably associated with adverse prognosis.59,60 However, the relationship between the depth of clonal clearance and recurrence has not been established in MDS.61 This represents a challenging (and costly) approach to serial sequencing and then perhaps sequential targeted therapies through a patient’s course.
Finally, it is important to note that genetic variability in MDS (and thus heterogeneous disease phenotypes) refers to the complexities of gene comutations as well as the diverse clonal architecture to these disorders.39,62 In some patients, a mutated gene may exist in a small subclone whereas in another it might represent an earlier event or mutation present in every tumor cell. Furthermore, this clonal architecture evolves over time and with treatment. The clinical implication of mutations must be considered in the context of other mutations as well as their associated clone size.34 Combination targeting with our established and novel therapeutics may ultimately be the key and is ever evolving; we will need to ensure we are not combining toxicities as well.
Conclusions
Genetic mutational profiling in MDS is increasing our understanding of the biology of the disease. This added knowledge is being used for possibly earlier diagnosis of the disease, more precise risk stratification, and in some instances, to guide potential therapies in a biologically rational way. The promise of effective, targeted therapies is real, if not yet realized. No new therapies have been approved for MDS in over a decade, yet this does not mean that the continued search for both targeted and novel therapeutics is a fool’s errand. Rather, as the pathobiology of MDS is further elucidated, our noble quest for effective and personalized treatment options for MDS patients inches closer to completion.
Correspondence
Amy E. DeZern, Division of Hematologic Malignancies; 1650 Orleans St, CRBI Rm 3M87, Baltimore, MD 21287-0013; e-mail: adezern1@jhmi.edu.
