
Abstract
Therapeutic options for acute lymphoblastic leukemia, especially in the relapsed/refractory setting, have expanded significantly in recent times. However, this comes at the cost of toxicities: medical as well as financial. We highlight some of the unique toxicities associated with the novel agents to apprise our readers about what to expect, how to recognize them, and how to manage these toxicities. One of the toxicities seen with inotuzumab, a CD22 antibody drug conjugate, is sinusoidal obstruction syndrome, which can be fatal in >80% of patients if associated with multiorgan failure. Blinatumomab, a monoclonal antibody targeting CD19, is associated with cytokine release syndrome (CRS) and neurotoxicity, both of which require prompt recognition and management primarily with corticosteroids. CRS and neurotoxicity are more common and more severe with chimeric antigen receptor T-cell therapy (CAR-T). The fact that CAR-T cannot be discontinued on demand adds a layer of complexity to the management of related toxicities of this therapy. Tocilizumab, an interleukin-6 receptor blocker, is used to treat severe CRS from CAR-T, whereas corticosteroids remain the mainstay for neurotoxicity management. Although effective, these drugs carry a high price tag, and we review the available data on cost-effectiveness of these agents, keeping in mind that median follow-up on most of these studies is limited and that long-term data on durability of response remain to be seen.
Learning Objectives
Learn about the risk and prevention of sinusoidal obstruction syndrome with inotuzumab, especially in patients who undergo allogeneic stem cell transplantation
Learn about recognition and management of cytokine release syndrome, and neurotoxicity associated with blinatumomab
Learn about potential toxicities of chimeric antigen receptor T-cell therapy in the form of cytokine release syndrome, neurotoxicity, and persistent hypogammaglobulinemia
Introduction
Relapsed/refractory acute lymphoblastic leukemia (ALL) has been associated with rather dismal prognosis, with 5-year overall survival reported to be <10% in older studies and 3-year overall survival reported as 24% in a more recent study.1-3 This mandates continued exploration of options for patients with newly diagnosed and relapsed/refractory ALL. Life-saving response with chimeric antigen receptor T-cell therapy (CAR-T) in the first patient treated and emergence of monoclonal antibody–based therapies led the way for the unfolding of a new era of novel agents in treatment of hematological malignancies. This bliss of incremental improvement in therapeutic options, however, has been fraught with fear of the accompanying toxicities and apparent high cost of these agents. In this review, we present data on appraisal and management of specific toxicities associated with each of the novel agents for management of ALL. We highlight to our readers the importance of prompt recognition and appropriate management of these toxicities. Additionally, we present available data on value-based care and the nuances related to its interpretation.
Inotuzumab ozogamicin
Inotuzumab ozogamicin is a humanized anti-CD22 antibody conjugated to calicheamicin. CD22 is a transmembrane sialoglycoprotein that is expressed on >90% of mature and precursor B cells, undergoes constitutive endocytosis, and is not shed into extracellular matrix.4-6 Calicheamicin is a cytotoxic natural product of Micromonospora echinospora that induces cell death in target cells by interactions with double-helical DNA.7,8 After binding to CD22, inotuzumab is internalized into lysosomes, where calicheamicin leads to double-strand DNA cleavage and subsequent apoptosis.8-10
A phase 2 trial administered inotuzumab initially at 1.8 mg/m2 every 3 to 4 weeks and subsequently, at 0.8 mg/m2 on day 1 followed by 0.5 mg/m2 on days 8 and 15 in monthly cycles.11,12 Responses were seen in 58% to 68% of patients in 2 early-phase studies for relapsed/refractory ALL.12,13 The phase 3 INO-VATE trial showed higher response rates (81% vs 29%) and higher minimal residual disease (MRD) negativity (78% vs 28%) with inotuzumab compared with standard therapy in patients with relapsed/refractory ALL.14 The median overall survival was 7.7 months for inotuzumab vs 6.7 months for standard therapy (P = .04), and in a post hoc restricted mean) survival time analysis, median overall survival was 13.9 vs 9.9 months, respectively(P = .0023). Based on these results, inotuzumab was approved by the Food and Drug Administration (FDA) in August 2017.
Unique and important toxicities related to inotuzumab
Hepatic toxicity, including sinusoidal obstruction syndrome.
With all studies utilizing inotuzumab, hepatic adverse events have emerged as a distinct toxicity. Of these, sinusoidal obstruction syndrome (SOS), also known as veno-occlusive disease, is of utmost clinical implication given the significant morbidity and reported fatality rate of over 80% in patients who develop multiorgan failure as a consequence.15 Rates and grades of development of hyperbilirubinemia, liver enzyme elevation, and SOS are tabulated in Table 1, with each study utilizing inotuzumab. Of note, the rate of SOS development after allogeneic stem cell transplantation (HCT) was lower in patients who received weekly inotuzumab compared with those who received single-dose inotuzumab in the initial phase 2 trial (7% and 23%, respectively).12 This may be related to use of a dual alkylator as a conditioning regimen more commonly in patients who received single-dose inotuzumab in addition to the potentially higher toxicity of a single-dose regimen. It is important to note that, despite initial favorable responses, inotuzumab therapy usually needs to be followed by HCT to maintain durability of the response, which increases the risk of SOS. For instance, in the INO-VATE study, of the 48 patients who underwent HCT after inotuzumab, 10 (21%) developed SOS, and 5 (10%) died consequently.14
Unique toxicities of novel agents for B-cell ALL
| Drug | Toxicity | Clinical trials | ||||||
|---|---|---|---|---|---|---|---|---|
| Inotuzumab | Phase 212 | Phase 1/213 | Phase 314 | |||||
| N = 90; median 39.5 y (range, 4-84 y) | N = 72; median 45 y (range, 20-79 y) | N = 109; median 47 y (range, 18-78 y) | ||||||
| Hyperbilirubinemia | Grade 1-2: 5% (w); 24% (sd) | Grade 1-2: 10% | Grade 1-2: 11% | |||||
| Grade ≥3: 0 (w); 41% (sd) | Grade ≥3: 0 | Grade ≥3: 4% | ||||||
| Liver enzyme elevation | Grade 1-2: 22% (w); 55% (sd) | Grade 1-2: 32% | Grade 1-2: 33% | |||||
| Grade ≥3: 5% (w); 2% (sd) | Grade ≥3: 6% | Grade ≥3: 8% | ||||||
| SOS | 7% (w) and 23% (sd) undergoing HCT | 8% undergoing HCT; 4% not undergoing HCT | 22% undergoing HCT; 8% not undergoing HCT | |||||
| Blinatumomab | Phase 235 | Phase 237 | Phase 236 | Phase 338 | Phase 239 | |||
| N = 21; median age 47 y (range, 20-77 y) | N = 36; median age 32 y (range, 18-77 y) | N = 189; median age 39 y (range, 18-79 y) | N = 267; mean age 40.8 y (range, 18-80 y) | N = 45; median age 55 y (range, 23-78 y) | ||||
| CRS | NA | Grade 1-2: NA | Grade 1-2: NA | Grade 1-2: 9% | Grade 1-2: 7% | |||
| Grade ≥3: 6% | Grade ≥3: 2% | Grade ≥3: 5% | Grade ≥3: 0 | |||||
| Neurotoxicity | 20% (grades not specified) | Grade 1-2: 22% | Grade 1-2: 39% | Grade 1-2: 36% | Grade 1-2: 44% | |||
| Grade ≥3: 14% | Grade ≥3: 13% | Grade ≥3: 9% | Grade ≥3: 7% | |||||
| Hypogammaglobulinemia | 24% | 8% | NA | 6% | NA | |||
| CAR-T | Phase 249 | Phase 150 | Phase 259 | Phase 151 | ||||
| N = 30; median age 14 y (range, 5-16 y) | N = 92 (treated =75); median age 11 y(range, 3-23 y) | N = 21; median age 13 y (range, 5-27 y) | N = 30; median age 39.5 y (range, 20-73 y) | N = 83 (treated =53); median age 44 y(range, 23-74 y) | ||||
| CRS | Grade 1-2: 73% | Grade 1-2: 43% | Grade 1-2: 48% | Grade 1-2: 60% | Grade 1-2: 59% | |||
| Grade ≥3: 27% | Grade ≥3: 36% | Grade ≥3: 28% | Severe: 23% (grade 5 = 3%) | Grade ≥3: 26% (grade 5 = 2%) | ||||
| Neurotoxicity | 43% (grades not specified) | Grade 1-2: 27% | Grade 1-2: 38% | Grade 1-2: 0 | Grade 2: 2% | |||
| Grade ≥3: 13% | Grade ≥3: 5% | Grade ≥3: 50% (grade 5 = 3%) | Grade ≥3: 42% | |||||
| Hypogammaglobulinemia | 90% (100% responders) | 81% (100% responders) | NA | NA | NA | |||
| Drug | Toxicity | Clinical trials | ||||||
|---|---|---|---|---|---|---|---|---|
| Inotuzumab | Phase 212 | Phase 1/213 | Phase 314 | |||||
| N = 90; median 39.5 y (range, 4-84 y) | N = 72; median 45 y (range, 20-79 y) | N = 109; median 47 y (range, 18-78 y) | ||||||
| Hyperbilirubinemia | Grade 1-2: 5% (w); 24% (sd) | Grade 1-2: 10% | Grade 1-2: 11% | |||||
| Grade ≥3: 0 (w); 41% (sd) | Grade ≥3: 0 | Grade ≥3: 4% | ||||||
| Liver enzyme elevation | Grade 1-2: 22% (w); 55% (sd) | Grade 1-2: 32% | Grade 1-2: 33% | |||||
| Grade ≥3: 5% (w); 2% (sd) | Grade ≥3: 6% | Grade ≥3: 8% | ||||||
| SOS | 7% (w) and 23% (sd) undergoing HCT | 8% undergoing HCT; 4% not undergoing HCT | 22% undergoing HCT; 8% not undergoing HCT | |||||
| Blinatumomab | Phase 235 | Phase 237 | Phase 236 | Phase 338 | Phase 239 | |||
| N = 21; median age 47 y (range, 20-77 y) | N = 36; median age 32 y (range, 18-77 y) | N = 189; median age 39 y (range, 18-79 y) | N = 267; mean age 40.8 y (range, 18-80 y) | N = 45; median age 55 y (range, 23-78 y) | ||||
| CRS | NA | Grade 1-2: NA | Grade 1-2: NA | Grade 1-2: 9% | Grade 1-2: 7% | |||
| Grade ≥3: 6% | Grade ≥3: 2% | Grade ≥3: 5% | Grade ≥3: 0 | |||||
| Neurotoxicity | 20% (grades not specified) | Grade 1-2: 22% | Grade 1-2: 39% | Grade 1-2: 36% | Grade 1-2: 44% | |||
| Grade ≥3: 14% | Grade ≥3: 13% | Grade ≥3: 9% | Grade ≥3: 7% | |||||
| Hypogammaglobulinemia | 24% | 8% | NA | 6% | NA | |||
| CAR-T | Phase 249 | Phase 150 | Phase 259 | Phase 151 | ||||
| N = 30; median age 14 y (range, 5-16 y) | N = 92 (treated =75); median age 11 y(range, 3-23 y) | N = 21; median age 13 y (range, 5-27 y) | N = 30; median age 39.5 y (range, 20-73 y) | N = 83 (treated =53); median age 44 y(range, 23-74 y) | ||||
| CRS | Grade 1-2: 73% | Grade 1-2: 43% | Grade 1-2: 48% | Grade 1-2: 60% | Grade 1-2: 59% | |||
| Grade ≥3: 27% | Grade ≥3: 36% | Grade ≥3: 28% | Severe: 23% (grade 5 = 3%) | Grade ≥3: 26% (grade 5 = 2%) | ||||
| Neurotoxicity | 43% (grades not specified) | Grade 1-2: 27% | Grade 1-2: 38% | Grade 1-2: 0 | Grade 2: 2% | |||
| Grade ≥3: 13% | Grade ≥3: 5% | Grade ≥3: 50% (grade 5 = 3%) | Grade ≥3: 42% | |||||
| Hypogammaglobulinemia | 90% (100% responders) | 81% (100% responders) | NA | NA | NA | |||
CRS, cytokine release syndrome; HCT, allogeneic stem cell transplantation; NA, not available; sd, single dose; w, weekly
What is SOS?
SOS is a potentially life-threatening complication that is triggered by injury to the hepatic endothelium and hepatocytes in zone 3 of hepatic acinus, resulting in penetration of red blood corpuscles into the space of Disse beneath the endothelial cells and obstructing the sinusoidal flow downstream.16-19 Eventually, progressive venular occlusion ensues and results in zonal liver damage and centrilobular hemorrhagic necrosis. The pathophysiology of SOS with inotuzumab is less well understood. However, given the similar risk seen with gemtuzumab, another antibody drug conjugate bound to calicheamicin, it is likely a direct effect from the calicheamicin.19,20
Who is at risk?
Risk factors for SOS outside inotuzumab use include use of myeloablative conditioning regimens with HCT (especially including busulfan or total body irradiation), second HCT, unrelated donor transplantation, older age, poorer Karnofsky performance score, and preexisting liver disease.18 Specific to the use of inotuzumab, use of 2 alkylating agents in the conditioning regimen, busulfan containing conditioning regimen, and prior elevation in bilirubin levels were associated with an increased risk of developing SOS.21 In the post hoc analysis of the INO-VATE data, SOS was higher in patients who underwent HCT after inotuzumab and were ≥55 years old compared with those who were younger (41% vs 17%).22
When and what to look out for.
In a setting of HCT, SOS typically develops within the first 21 days after HCT, although late-onset SOS has been reported.23-25 Similar results were seen in inotuzumab trials. Combined data from the INO-VATE and B1931010 trials showed that 7 patients developed SOS during or shortly after treatment with inotuzumab (without HCT) after administration of a median of 3 cycles (range, 1-6 cycles) at a median of 16 days (range, 8-60 days) from the last dose of inotuzumab.21 Of the patients who underwent a subsequent HCT, 19 of 101 (19%) developed SOS after a median of 3 cycles (range, 1-5) of treatment, after a median of 15 days (range, 3-57) from HCT, and after a median of 36 days (range, 16-134) from last inotuzumab therapy.21
Clinical suspicion of SOS should arise in patients with elevated bilirubin after HCT who present with weight gain >5% above baseline due to fluid retention, hepatomegaly, or right upper quadrant pain and/or ascites.17,18,24 Ultrasound can further identify hepatomegaly and ascites as well as decrease in velocity or reversal of portal flow. Transjugular liver biopsy, although the diagnostic gold standard, remains limited by its invasive approach, frequent association of thrombocytopenia with SOS, and lack of diagnostic utility.26,27
Prevention and management.
Because many patients treated with inotuzumab will undergo HCT, additional preventative measures are warranted to mitigate the risk of SOS. Recommendations from an expert panel include strategies avoiding the above described risk factors, such as use of conditioning regimens containing dual alkylating agents, and avoiding >2 cycles of inotuzumab before HCT among others as listed in Table 2.28
Sinusoidal obstructive syndrome associated with inotuzumab
| Diagnosis (new EBMT criteria18 ) | Prevention | Treatment |
|---|---|---|
| Within 21 d from HCT | Avoid double alkylator for conditioning regimen with HCT | Permanent discontinuation of inotuzumab if occurs while on therapy |
| Bilirubin ≥2 mg/dL along with 2 of the following: (1) painful hepatomegaly, (2) weight gain >5%, and (3) ascites | Avoid treatment with >2 cycles of inotuzumab if planning HCT after induction | Supportive therapy for fluid balance and pain control |
| Late onset >21 d after HCT | Avoid concomitant hepatotoxic medication use (eg, azoles) | Paracentesis if respiratory compromise due to ascites |
| Symptoms as the criteria above | Encourage use of prophylactic agents, such as ursodiol during HCT | Limit fluid removal with paracentesis to <1 L to avoid disruption of renal perfusion |
| Histological diagnosis of SOS | Defibrotide for severe SOS | |
| Two of the above criteria along with hemodynamic and/or ultrasound evidence of SOS |
| Diagnosis (new EBMT criteria18 ) | Prevention | Treatment |
|---|---|---|
| Within 21 d from HCT | Avoid double alkylator for conditioning regimen with HCT | Permanent discontinuation of inotuzumab if occurs while on therapy |
| Bilirubin ≥2 mg/dL along with 2 of the following: (1) painful hepatomegaly, (2) weight gain >5%, and (3) ascites | Avoid treatment with >2 cycles of inotuzumab if planning HCT after induction | Supportive therapy for fluid balance and pain control |
| Late onset >21 d after HCT | Avoid concomitant hepatotoxic medication use (eg, azoles) | Paracentesis if respiratory compromise due to ascites |
| Symptoms as the criteria above | Encourage use of prophylactic agents, such as ursodiol during HCT | Limit fluid removal with paracentesis to <1 L to avoid disruption of renal perfusion |
| Histological diagnosis of SOS | Defibrotide for severe SOS | |
| Two of the above criteria along with hemodynamic and/or ultrasound evidence of SOS |
EBMT, European Society for Blood and Marrow Transplantation; HCT, allogeneic stem cell transplantation.
Supportive measures for patients with suspected SOS are tabulated in Table 2. Defibrotide use is recommended in patients with severe SOS and those with renal or pulmonary dysfunction.29 Of 26 patients who developed SOS after inotuzumab therapy in INO-VATE and B1931010 studies, defibrotide was used in 14 patients; SOS resolved in 6 (43%), persisted in 5 (36%), and was fatal in another 3 (21%) patients.21 Although these responses are encouraging, there is room for additional improvement in management of SOS, especially because the absolute number of patients treated is relatively low. Prompt recognition and appropriate management of SOS remain imperative. The use of defibrotide in a prophylaxis setting is currently under investigation for patients at high risk of developing SOS, and it is not recommended outside the setting of a clinical trial.
Financial reflection and cost considerations
In the United States, the cost of inotuzumab is around $89 760 for the first cycle, $67 320 for subsequent cycles for patients in complete remission, and $89 760 for subsequent cycles for patients not in complete remission after the first cycle (based on an average body surface area of 1.7 m2). For patients going to HCT, 2 cycles of treatment are recommended, and for patients who are not candidates for HCT, up to 6 cycles can be used.
Data from the INO-VATE trial were used in determining the life years (LYs) and quality-adjusted life years (QALYs) gained with the use of inotuzumab compared with standard therapy. Using a Markov model, there was an overall increment of 2.34 LYs and 1.81 QALYs with the use of inotuzumab compared with standard therapy.30 The maximum increment in LYs and QALYs was seen post-HCT. Subsequently, the United Kingdom National Institute for Health and Care Excellence conducted a cost-effectiveness analysis, which calculated a deterministic incremental cost-effectiveness ratio (ICER) of £114.078 per QALY gained (∼$150 000).31 Historically, in the United States, an ICER threshold of $50 000 per QALY has been deemed appropriate to discern that an intervention is cost effective, but this was established >3 decades ago. Given the current financial situation, an ICER threshold of $150 000 to $300 000 per QALY for oncology has been proposed.32,33 Per the new proposed benchmark, which seems more acceptable accounting for current day expenses, the ICER for inotuzumab falls at the cusp of being cost effective.
However, there are several limitations to the cost analysis that was put forth. First, the follow-up on the INO-VATE trial is relatively short, and hence, long-term benefit of the drug remains to be seen. Second, this analysis does not include the cost associated with subsequent health care utilization and interventions, such as HCT or management of SOS, that can be attributed to the use of inotuzumab. Third, because the benefit was more pronounced in patients who underwent HCT after inotuzumab therapy, the benefit of inotuzumab in patients not eligible for HCT and the cost implications thereof remain unclear.
Nevertheless, it is important to recognize that relapsed/refractory ALL is an indication that has otherwise had dismal outcomes as indicated above. Hence, an effective drug at an affordable price is an unmet need.
Blinatumomab
With a bispecific T-cell engager antibody construct, blinatumomab has dual binding sites: one for CD3-positive cytotoxic T cells and another for CD19-positive ALL blasts. On binding to the anti-CD3 arm of blinatumomab, T cells get activated and induce perforin-mediated lysis via granzyme entry into target ALL cells with subsequent apoptosis.34
Results from an initial pilot trial and 2 subsequent phase 2 trials showed complete response in 43% to 69%, with negative MRD achieved in ≥80%.35-37 A subsequent phase 3 multi-institutional trial showed a higher rate of complete remission (34% vs 16%, P < .001) and superior median overall survival with blinatumomab compared with standard therapy (7.7 vs 4 months, P = .01).38 In patients with Philadelphia chromosome–positive ALL, a complete response was seen in 16 of 45(36%) patients on treatment with blinatumomab in a phase 2 study.39 Consequently, blinatumomab is now approved for Philadelphia-negative and -positive relapsed/refractory ALL as well as treatment of patients with ALL who are in remission but MRD positive.
Unique and important toxicities related to blinatumomab
Cytokine release syndrome.
Cytokine release syndrome (CRS) is one of the notable toxicities of blinatumomab, and along with neurotoxicity, it is included as a boxed warning. The rates of CRS observed in each study are tabulated in Table 1. In the phase 2 and phase 3 studies, prephase dexamethasone was instituted in patients with high disease burden along with stepwise escalation of dose in cycle 1 (9 µg/d for 1 week followed by 28 µg/d).14,36 This was established in an earlier study where 2 patients with high leukemia burden developed grade 4 CRS prior to introduction of stepwise dose escalation and prephase dexamethasone.37
What is CRS with blinatumomab?
CRS refers to a systemic inflammatory response resulting from antigen-antibody interactions when using monoclonal antibodies that leads to activation of cytotoxic T cells that release inflammatory cytokines. The exact mechanism remains unclear, but elevation in interleukin-6 (IL-6), IL-10, and interferon-γ has been noted in patients receiving blinatumomab for ALL.40 It has also been hypothesized that a mechanism similar to hemophagocytic lymphohistiocytosis or macrophage activation syndrome may be responsible for blinatumomab-related CRS.41
Who is at risk?
The 2 patients who developed grade 4 CRS in the earlier study were started on full-dose blinatumomab and had high leukemia burden (88% and 90% blasts) in addition to extramedullary ALL involvement in 1 patient.37 Of note, both of these patients were showing good response to blinatumomab therapy in contrast with nonresponders, none of whom had CRS. This infers that a higher disease burden and a higher initial starting dose of infusion are associated with a higher risk of CRS.
When and what to look out for.
CRS is primarily seen with the first cycle of blinatumomab. Clinical presentation may be mild and limited to pyrexia, malaise, headache, or nausea, but it can escalate to hypotension, hypoxia, renal impairment, and elevation in transaminases or bilirubin and even be life threatening with pulmonary edema, capillary leak syndrome, or rarely, disseminated intravascular coagulopathy or hemophagocytic lymphohistiocytosis.36,41,42 It is important to grade CRS to enable formulation of a treatment plan based on severity (Table 3). The National Cancer Institute (NCI) Common Terminology Criteria for Adverse Effects (CTCAE) has developed a specific grading scale for CRS that is different from grading of standard immune reactions as tabulated below.
National Cancer Institute Common Terminology Criteria for Adverse Events grading and management of CRS from blinatumomab
| Grade | Criteria* | Management† |
|---|---|---|
| Grade 1 | Fever ± constitutional symptoms | Symptom management without interruption of therapy |
| Grade 2 | Hypotension not requiring pressors, responding to fluids | Symptomatic treatment with intravenous fluids, respiratory support, anti-inflammatory, narcotics; interrupting blinatumomab can be considered |
| Hypoxia responsive to <40% O2 | ||
| Grade 3 | Hypotension managed with one pressor | Discontinue blinatumomab until resolution; resume at 9 µg/d and then escalate to 28 µg/d if recurrence of CRS after 7 d |
| Hypoxia requiring ≥40% O2 | ||
| Grade 4 | Life-threatening complications | Discontinue blinatumomab permanently; if refractory to corticosteroids, tocilizumab may be considered |
| Urgent intervention indicated | ||
| Grade 5 | Death | — |
| Grade | Criteria* | Management† |
|---|---|---|
| Grade 1 | Fever ± constitutional symptoms | Symptom management without interruption of therapy |
| Grade 2 | Hypotension not requiring pressors, responding to fluids | Symptomatic treatment with intravenous fluids, respiratory support, anti-inflammatory, narcotics; interrupting blinatumomab can be considered |
| Hypoxia responsive to <40% O2 | ||
| Grade 3 | Hypotension managed with one pressor | Discontinue blinatumomab until resolution; resume at 9 µg/d and then escalate to 28 µg/d if recurrence of CRS after 7 d |
| Hypoxia requiring ≥40% O2 | ||
| Grade 4 | Life-threatening complications | Discontinue blinatumomab permanently; if refractory to corticosteroids, tocilizumab may be considered |
| Urgent intervention indicated | ||
| Grade 5 | Death | — |
Adapted from Common Terminology Criteria for Adverse Events, Version 5.0, November 2017, National Institutes of Health.
Adapted from blinatumomab (BLINCYTO) packaging insert.
Prevention and management.
As noted above, prevention strategies with prephase dexamethasone and stepwise increase in blinatumomab dosing should be used.35,36,38 Additionally, dexamethasone use at the time of dose step up (on day 8 of cycle 1) and when resuming therapy after interruption of 4 hours has been recommended. The therapeutic mainstay is corticosteroids and temporary interruption or permanent discontinuation of treatment based on the severity or grading of CRS (Table 3). Permanent discontinuation should be avoided by appropriate management of clinical features of toxicity in possible cases. In a report of 1 patient who developed hemophagocytic lymphohistiocytosis with blinatumomab, management with tocilizumab, an IL-6 receptor blocker, was successful without mitigating the efficacy of blinatumomab.41
Neurotoxicity.
A wide spectrum of neurotoxicity was seen in the clinical trials of blinatumomab, making this one of the most common reasons for interruption of therapy. Overall rates from all studies are shown in Table 1. In the larger phase 2 trial, no grade 5 neurotoxicity was observed, and all patients, with the exception of 3 who died of unrelated causes, subsequently recovered from the neurological event.36 A total of 29 (15%) patients had drug interruptions due to neurotoxicity. In the phase 3 study, neurotoxicity led to drug interruptions in 6% of patients and drug discontinuation in 4% of patients.38
What is neurotoxicity from blinatumomab?
Neurotoxicity attributed to blinatumomab can range from headache, malaise, confusion, somnolence, or disorientation to more ominous forms, such as ataxia, seizure, aphasia, and stupor. Due to a similar occurrence with CAR-T, one plausible mechanism proposed is disruption of the blood-brain barrier by activated T cells and cytokine release on binding to CD19-positive B cells in the central nervous system (CNS).43 Data suggest variable expression of CD19 in the CNS, which may explain the occurrence of neurotoxicity only in a subset of patients.44
Who is at risk?
It is currently not well defined who would be at a higher risk. Most of these trials excluded patients with clinically relevant CNS disease, including CNS involvement with ALL, because this was fraught with a plausible higher risk for neurotoxicity and inability to differentiate CNS disease progression from neurotoxicity. Nonetheless, it is reasonable to believe that patients with CNS involvement with ALL may be at a higher risk of developing neurotoxicity.
When and what to look out for.
Prevention and management.
Prophylaxis with antiepileptics is not indicated currently, because the incidence of seizure development is low, being <1% in the phase 3 trial.38 Drug discontinuation and treatment with dexamethasone are recommended for grade ≥3 neurological events, but the latter could be considered sooner to avoid severe toxicities.42 For patients with grade 3 CNS adverse events, reintroduction of blinatumomab can be considered after the preexisting neurological toxicity has improved to at least grade 1 level for at least 3 days, with resumption at lower dose of 9 μg/d.42 In patients with grade 4 toxicity or those in whom grade 3 toxicity lasts >7 days or recurs on reintroduction, permanent discontinuation is mandated.
Hypogammaglobulinemia.
Financial reflection and cost considerations
The cost of blinatumomab amounts to around $89 000 per cycle in the United States, not including the hospitalization for 9 days for the first cycle and 2 days for the second cycle. A median of 2 cycles was used in the TOWER trial (range, 1-9).38
Cost-effectiveness analysis for blinatumomab was done using data from the TOWER trial using a partitioned survival model.38,46 The standard of care arm in this study used fludarabine/high-dose arabinoside, high-dose arabinoside alone, or high-dose methotrexate or clofarabine regimens, all of which have previously shown relatively poor outcomes in these patients. For an incremental cost of $180 642 with blinatumomab compared with standard therapy, largely related to greater treatment cost associated with blinatumomab and partially offset by reduced cost of subsequent salvage therapies, there was an increment of 1.92 LYs and 1.64 QALYs, amounting to an ICER of $110 108 per QALY gained.46 These projections were made with extrapolation of data from the TOWER study due to nonavailability of long-term data.
This cost-effectiveness analysis, like many others, is limited by arbitrary assumptions, short follow-up in studies, and not including the cost of subsequent treatments that might be warranted. The quality of life data in the analysis were assumed to be similar to those of the age-matched population at 4 years, which again, is oblivious to the long-term toxicities incurred by the treatment or disease. Additionally, although blinatumomab was used for maintenance in the trial, it was not included in the prescribing information. A scenario analysis was done to overcome this by setting the drug acquisition cost to 0 after the initial 5 cycles (calculated ICER = $98 917 per QALY gained); however, this remains riddled with the uncertainty of loss of clinical benefit without the use of maintenance in this scenario.
Considering various limitations, the drug does seem to clearly benefit patients with B-cell ALL, especially those with low-volume disease, but its cost-effectiveness remains uncertain given the limitations of the financial analyses done to date.
CAR-T
CAR-T has evolved as a promising adoptive T-cell therapy, and it was deservingly labeled by the American Society of Clinical Oncology as the “Advance of the Year” for 2018. CAR-T is genetically engineered to express a receptor that couples an anti-CD19 single-chain Fv domain to intracellular T-cell signaling domains. Tisagenlecleucel (previously CTL019) was approved by the FDA for treatment of relapsed/refractory B-cell ALL in patients ≤25 years old. After initial series,47 subsequent reports showed complete response in 90% of patients in a single center and 83% of patients in the multinational ELIANA trial.48,49 Overall survival was 90% at 6 months and 76% at 12 months in the latter.49 CAR-T data from Memorial Sloan-Kettering Cancer Center (MSKCC) and the NCI also show a response rate in the range of 70% to 90%, with differences in chimeric antigen receptor construct.50,51 Despite the deserving applause to CAR-T, there remain on-target/off-tumor and off-target antigen recognition nuances about the therapy that warrant attention for safe clinical application. Currently, the availability to administer tisagenlecleucel is restricted under a risk evaluation and mitigation strategy at select centers to allow for use only by centers with expertise in management of toxicities.
Unique and important toxicities related to CAR-T
CRS.
The most feared adverse events noted with CAR-T are CRS and neuropsychiatric toxicity (described below). As depicted in Table 1, the variability in rates of CRS in different trials has been attributed to the differences in the construct, the age groups treated, and the clinical practices at the various centers. Also, CRS with CAR-T (grade ≥3 = 26%-46%) is more frequent and potentially more severe compared with that reported with blinatumomab (grade ≥3 in ≤5%).38
What is CRS with CAR-T?
CRS with CAR-T results from immune activation and subsequent elevated inflammatory cytokines. Remarkable increases in cytokines, such as IL-6, IL-10, interferon-γ, granulocyte-macrophage colony-stimulating factor, and more recently, host macrophage–derived IL-1, IL-6, and nitric oxide, have been indicted for the CRS after CAR-T infusion (Figure 1).48,50,52-54
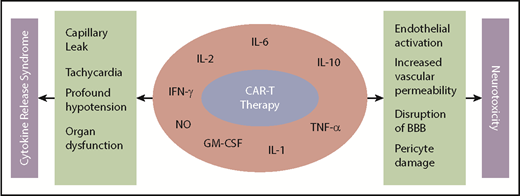
Mechanism of CRS and neurotoxicity with CAR-T. BBB, blood-brain barrier; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon-γ; NO, nitric oxide; TNF-α, tumor necrosis factor-α.
Mechanism of CRS and neurotoxicity with CAR-T. BBB, blood-brain barrier; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon-γ; NO, nitric oxide; TNF-α, tumor necrosis factor-α.
Who is at risk?
A mild form of CRS was seen in a majority of patients treated with CAR-T. A higher disease burden seemed to drive a more severe CRS.49-51 The rate of CRS seen with the 19-41BB construct (University of Pennsylvania [UPenn]) was higher than that seen with the 19-28z construct (MSKCC) in their respective studies.49,51 Hence, it is plausible that the construct of the chimeric receptor in addition to other factors, such as prior therapies and degree of lymphodepletion, may influence the rate and severity of CRS.55 This, however, should be interpreted in light of the fact that these were 2 different trials done in different clinical settings, where practice variability might also account for these differences.
When and what to look out for.
Unlike blinatumomb, where CRS occurs most commonly with the initial infusion, CRS with CAR-T can occur immediately after or be a delayed response occurring days or weeks after infusion depending on the kinetics of T-cell expansion. The earlier reports showed that most CRS occurred in the first 2 weeks, with rare reports beyond 17 days.48,56 In the ELIANA trial, episodes of grade ≥3 CRS occurred in patients up to 8 weeks after infusion.49 Patients usually present with high fever, malaise, fatigue, myalgias, tachycardia, hypotension, respiratory distress, capillary leak, and organ failure. Respective teams at UPenn and the NCI described criteria for grading CRS that are modified from the CTCAE system to account for differences in CRS with CAR-T compared with other therapies, the possibility of delayed CRS and inability to discontinue or “turn-off” the CAR-T effect (Table 4). Additionally, biomarkers are being explored to identify patients who would be at a higher risk of developing severe CRS.57 We currently use C-reactive protein and ferritin levels as biomarkers for monitoring patients after CAR-T infusion, because these are the 2 commercially available laboratory tests available that have been correlated with development and severity of CRS among various studies.49,52,58,59 Clinical presentation, however, remains the mainstay for suspicion, because these levels can sometimes lag behind or may have variability in peak levels.
Grading and management of CRS from CAR-T
| Grade | UPenn criteria55,56 | NCI criteria67 | Management |
|---|---|---|---|
| Grade 1 | Mild reaction treated with supportive care, such as antipyretics, antiemetics | Nonlife-threatening symptoms that require symptomatic management | Vigilant supportive management |
| For example, fever, nausea, fatigue, headache, myalgias, malaise | Antipyretics, analgesics | ||
| Monitor fluid balance | |||
| Rule out infections | |||
| Grade 2 | Moderate reaction with signs of organ dysfunction related to CRS, not attributable to other etiologies (eg, grade 2 creatinine or grade 3 LFT elevation) | Symptoms require and respond to moderate intervention | No extensive comorbidities or older age |
| Hospitalization for management of CRS-related symptoms, including fevers with associated neutropenia or need for intravenous therapies (other than intravenous fluids for hypotension) | Oxygen requirement <40% or hypotension responsive to fluids or low dose of one vasopressor | Vigilant supportive management as above; extensive comorbidities or older age | |
| Vigilant supportive care | |||
| Grade 2 organ toxicity | + Tocilizumab | ||
| ± Corticosteroids | |||
| Grade 3 | More severe reaction | Symptoms require and respond to aggressive intervention | Vigilant supportive care as above |
| Hospitalization required for organ dysfunction, including grade 4 LFT elevation or grade 3 creatinine elevation related to CRS and not attributable to any other conditions (excludes management of fever or myalgias but includes hypotension treated with intravenous fluids defined as multiple fluid boluses for blood pressure support or low-dose vasopressors) | Oxygen requirement ≥40% | Vasopressors as needed | |
| Coagulopathy requiring fresh frozen plasma, cryoprecipitate, or fibrinogen concentrate | Hypotension requiring high-dose or multiple vasopressors | + Tocilizumab | |
| Hypoxia requiring supplemental oxygen (nasal cannula oxygen, high-flow oxygen, CPAP, or BiPAP) | Grade 3 organ toxicity or grade 4 elevation in transaminases | ± Corticosteroids | |
| Patients admitted for management of suspected infection due to fevers and/or neutropenia may have grade 2 CRS | |||
| Grade 4 | Life-threatening complications | Life-threatening symptoms | Vigilant supportive care as above |
| Hypotension requiring high-dose vasopressors | Requiring ventilator support | Vasopressors as needed | |
| Hypoxia requiring mechanical ventilation | Grade 4 organ toxicity other than transaminase elevation | +Tocilizumab | |
| ± Corticosteroids | |||
| Grade 5 | Death | Death |
| Grade | UPenn criteria55,56 | NCI criteria67 | Management |
|---|---|---|---|
| Grade 1 | Mild reaction treated with supportive care, such as antipyretics, antiemetics | Nonlife-threatening symptoms that require symptomatic management | Vigilant supportive management |
| For example, fever, nausea, fatigue, headache, myalgias, malaise | Antipyretics, analgesics | ||
| Monitor fluid balance | |||
| Rule out infections | |||
| Grade 2 | Moderate reaction with signs of organ dysfunction related to CRS, not attributable to other etiologies (eg, grade 2 creatinine or grade 3 LFT elevation) | Symptoms require and respond to moderate intervention | No extensive comorbidities or older age |
| Hospitalization for management of CRS-related symptoms, including fevers with associated neutropenia or need for intravenous therapies (other than intravenous fluids for hypotension) | Oxygen requirement <40% or hypotension responsive to fluids or low dose of one vasopressor | Vigilant supportive management as above; extensive comorbidities or older age | |
| Vigilant supportive care | |||
| Grade 2 organ toxicity | + Tocilizumab | ||
| ± Corticosteroids | |||
| Grade 3 | More severe reaction | Symptoms require and respond to aggressive intervention | Vigilant supportive care as above |
| Hospitalization required for organ dysfunction, including grade 4 LFT elevation or grade 3 creatinine elevation related to CRS and not attributable to any other conditions (excludes management of fever or myalgias but includes hypotension treated with intravenous fluids defined as multiple fluid boluses for blood pressure support or low-dose vasopressors) | Oxygen requirement ≥40% | Vasopressors as needed | |
| Coagulopathy requiring fresh frozen plasma, cryoprecipitate, or fibrinogen concentrate | Hypotension requiring high-dose or multiple vasopressors | + Tocilizumab | |
| Hypoxia requiring supplemental oxygen (nasal cannula oxygen, high-flow oxygen, CPAP, or BiPAP) | Grade 3 organ toxicity or grade 4 elevation in transaminases | ± Corticosteroids | |
| Patients admitted for management of suspected infection due to fevers and/or neutropenia may have grade 2 CRS | |||
| Grade 4 | Life-threatening complications | Life-threatening symptoms | Vigilant supportive care as above |
| Hypotension requiring high-dose vasopressors | Requiring ventilator support | Vasopressors as needed | |
| Hypoxia requiring mechanical ventilation | Grade 4 organ toxicity other than transaminase elevation | +Tocilizumab | |
| ± Corticosteroids | |||
| Grade 5 | Death | Death |
BiPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; LFT, liver function tests.
Prevention and management.
Because higher disease burden has been correlated with increased CRS, pre–CAR-T cytoreduction is an option, but it remains to be formally studied. In addition to supportive management, IL-6 blockade was thought to be a feasible strategy when IL-6 was identified to be elevated 1000 times in the first patient with ALL who developed CRS from CAR-T. Tocilizumab was, therefore, FDA approved along with tisagenlecleucel for management of severe or life-threatening CRS. Corticosteroids can abate the inflammatory response of CRS; however, concerns about simultaneously mitigating antileukemic activity of CAR-T have been raised.52 Siltuximab, a direct IL-6 inhibitor, has been hypothesized as being potentially more effective in controlling CRS, because it would eliminate the concern for increased diffusion of IL-6 into the CNS, such as might occur with tocilizumab, which blocks the IL-6 receptor and results in an increase in IL-6 levels.60 Concepts, such as introduction of the “suicide gene” in the chimeric antigen receptor construct or use of CRISPR (clustered regularly interspaced short palindromic repeats)–modified T cells or antibody-dependent cell ablation, are being studied.61 Tabulation of treatment options based on CRS severity is provided in Table 4.
Neurotoxicity.
Like CRS, neuropsychiatric events have been more common and severe with CAR-T compared with blinatumomab. Neurotoxicity presenting as cerebral edema in 5 patients was the reason for termination of the pivotal phase 2 trial by Juno Therapeutics using JCAR015 in adult patients with B-cell ALL.62,63 Rates of neurotoxicity in various trials of different CAR-T products in B-cell ALL are shown in Table 1.
What is neurotoxicity from CAR-T?
Although the precise mechanism remains to be elucidated, data suggest endothelial activation and increased blood-brain barrier permeability resulting in high cytokine concentration in cerebrospinal fluid.64 This results in additional endothelial cell and pericyte activation, which if severe, can lead to cerebral edema or other manifestation of neurotoxicity from CAR-T (Figure 1). Mediators of neurotoxicity have been shown to be IL-1 and IL-6 derived from host macrophages as opposed to previously thought T cell–derived mediators.54
Who is at risk?
Like CRS, both higher disease burden and higher peak CAR-T expansion correlate with severe neurotoxic effects of CAR-T.51 Other factors that can lead to higher CAR-T expansion, such as higher dose of CAR-T or high-intensity lymphodepleting therapy, can also potentially increase the risk of neurotoxicity.
When and what to look out for.
Like neurotoxicity with blinatumomab, neurotoxicity manifestations from CAR-T include a similar constellation of neurological symptoms. Early signs can include inattention, impaired handwriting, and speech disturbances. More severe forms include seizure, obtundation, or cerebral edema. These occurred within days after infusion of CAR-T in most studies, but they could happen concurrent with, after resolution of, or without a co-occurrence of CRS.49,51 Predictive models using biomarkers (such as C-reactive protein, ferritin, monocyte chemoattractant protein-1, macrophage inflammatory protein 1α, interferon-γ, and IL-13) and/or baseline patient characteristics, such as platelet count, disease burden, and mean corpuscular hemoglobin concentration, have been studied in various studies with varying sensitivities and predictive abilities.57,58,65
Prevention and management.
Early recognition is key! Preventive measures, such as debulking chemotherapy in patients with high disease burden and using a lower dose range of CAR-T, have been proposed but warrant formal studies.66 Periodic neurological assessment and clinical monitoring for changes in mental status are the most important tools to facilitate early detection of onset of neurotoxicity. Management includes supportive care (such as use of analgesics and antiepileptic drugs), investigations for other potential causes (computed tomography scan, vitamin B1 deficiency, and electroencephalography), and comanagement with neurology colleagues. Corticosteroids are the mainstay in the absence of CRS, whereas tocilizumab is recommended if there is concurrent CRS.60 If mental status changes persist or worsening headache is noted, assessment of intracranial tension (fundoscopy) is recommended. If raised intracranial pressure is noted on imaging or with high opening pressure on lumbar puncture, high-dose corticosteroids, elevation of the head end of the bed, hyperosmolar therapy with mannitol or hypertonic saline, and removal of cerebrospinal fluid if ommaya catheter is available can be considered. Aspiration precautions and intubation, if mental status precludes maintaining airway, must be considered.
B-cell aplasia and hypogammaglobulinemia.
B-cell aplasia is an example of “on-target, off-tumor” activity of CAR-T cells, because CD19 is also expressed on normal B lymphocytes. B-cell aplasia resulting from CAR-T results in prolonged hypogammaglobulinemia. In the UPenn cohort, all responding patients developed B-cell aplasia.49 Thus, regular intravenous immunoglobulin replacement is needed for the duration of B-cell aplasia.
Financial reflection and cost considerations
As the success stories of CAR-T emerge, there is the customary concern about cost, because the price of tisagenlecleucel is set at $475 000. There is an outcomes-based agreement with Novartis, which calls for payment only for patients who get a morphological remission within a month of receiving treatment (>80% in the clinical trials). Whether response at 1 month is a reasonable timeframe for response remains to be seen, with durability of initial responses on longer follow-up. Also, the price does not include the cost of leukapheresis, administration costs, and the hospital cost incurred for management of the infusion or related side effects, including long-term (potentially lifelong) use of intravenous immunoglobulins. Cost-effectiveness analysis was conducted by the Institute of Clinical and Economics Review using the semi-Markov–partitioned survival model.31 Based on the analysis from the Institute for Clinical and Economic Review using data from the ELIANA trial for tisagenlecleucel and using a cost discount at 3% per year due to inflation, for an incremental cost of $329 498 compared with clofarabine, 7.91 incremental LYs and 7.18 incremental QALYs were gained. The calculated ICER is $41 642 per LY and $45 871 per QALY for CAR-T compared with clofarabine, which is lower than the historic benchmark of $50 000 per QALY and within the more recently suggested threshold of $150 000 for oncology therapies.33
Several limitations of this analysis need to be recognized. First, a major limitation is the use of clofarabine as a comparator, especially because more effective agents, such as blinatumomab and inotuzumab, have now been approved for this indication. Second, the nonavailability of randomized studies of CAR-T in B-cell ALL makes it challenging to accurately establish the place of CAR-T in the therapeutic paradigm for B-cell ALL amid other options, such as other novel agents as well as HCT, especially in light of cost considerations. Third, whether HCT would be required after CAR-T remains a matter of investigation, and the cost implications of that have not been included in this analysis. Fourth, response rates in various CAR-T trials are reported in patients who were treated per protocol and not as an intention to treat. Although still encouraging, an intention-to-treat analysis will dilute the apparent benefit from CAR-T. Fifth, the lack of long-term follow-up on most of these studies is a matter of concern from the standpoint of durability of response as well as late toxicities.
Cost justification for CAR-T is a seemingly never-ending debate, and the unprecedented price tag mitigates the scientific and medical enthusiasm for the therapy. Whether competing products and use of off-the-shelf CAR-T products will mitigate costs in the future remains to be seen.
Conclusions
The landscape of treatment of relapsed/refractory B-cell ALL has evolved dramatically and will continue to do so. To make these advances safe and effective, awareness of the potential complications as well as the successes is imperative. As use of these agents continues to expand and immunotherapies, like CAR-T, are applied to other disease settings, increased awareness of some of the unique toxicities of these agents will be important so that they are managed effectively to optimize patient outcomes. A word of caution while interpreting the cost analysis data is that B-cell ALL, especially in its relapsed/refractory form, has been a deadly disease for several decades. The availability and utilization of novel agents, although with limited follow-up, show promise in treatment of this otherwise deadly disease.
Correspondence
Mark R. Litzow, Division of Hematology and Bone Marrow Transplant, Mayo Clinic, 200 1st St SW, Rochester, MN 55905; e-mail: litzow.mark@mayo.edu.
This article was selected by the Blood Advances and Hematology 2018 American Society of Hematology Education Program editors for concurrent submission to Blood Advances and Hematology 2018. It is reprinted from Blood Advances 2018, Volume 2.
Off-label drug use: None disclosed.
