
Abstract
Allogeneic hematopoietic cell transplantation is a potentially curative treatment of different hematological malignancies. A major life-threatening complication is acute graft-versus-host disease (GVHD), in particular when the disease becomes steroid refractory. Based on the detection of pathogenic cytokines, chemokines, and T-cell subsets in individuals developing GVHD or experimental GVHD models, different therapeutic strategies have been developed. A potential cause why targeting individual receptors can lack efficacy could be that multiple cytokines, danger signals, and chemokine that have redundant functions are released during GVHD. To overcome this redundancy, novel strategies that do not target individual surface molecules like chemokine receptors, integrins, and cytokine receptors, but instead inhibit signaling pathways downstream of these molecules, have been tested in preclinical GVHD models and are currently being tested in clinical GVHD trials. Another important development is tissue regenerative approaches that promote healing of GVHD-related tissue damage as well as strategies that rely on microbiota modifications. These approaches are promising because they act very differently from conventional immunosuppression, instead aiming at reinstalling tissue homeostasis and microbiome diversity. This review discusses major novel developments in GVHD therapy that are based on a better understanding of GVHD biology, the repurposing of novel kinase inhibitors, microbiome modification strategies, and tissue-regenerative approaches.
Learning Objectives
Understand novel strategies that inhibit signaling pathways that promote GVHD
Define tissue-regenerative approaches that promote healing of GVHD-related tissue damage in thymus, intestinal tract, and skin, and microbiota modification that hold promise to extend immunosuppression-based acute GVHD therapy approaches
Introduction
The classical prophylaxis against acute and chronic graft-versus-host disease (GVHD) includes a calcineurin inhibitor (tacrolimus or cyclosporine A) and an antimetabolite (methotrexate or mycophenolate mofetil) (reviewed in Zeiser and Blazar1,2 ). The question as to whether the addition of a third immunosuppressive agent for patients with an unrelated donor would further reduce GVHD was studied in 2 randomized, multicenter phase 3 trials of rabbit anti-thymocyte globulin (ATG).3,4 The ATG trial performed in Europe showed decreased acute and chronic GVHD incidence without increased relapse or nonrelapse mortality when ATG was added to standard prophylaxis.3 A multicenter, prospective, placebo-controlled, double-blind, randomized clinical trial performed in North America showed that prophylactic use of ATG was also connected to reduced risk of acute and chronic GVHD, but also to a lower overall survival at 2 years, which was 74% in the placebo group and 59% in the group receiving ATG.4 The discrepant results with respect to survival, but not GVHD risk, of the 2 studies could be explained by multiple factors. One factor could be that patients who received ATG and underwent lymphocyte-depleting radiation had unusually unfavorable outcomes.4 This could be explained by low lymphocyte counts, which may have indirectly led to higher ATG levels, increasing both infectious complications and toxicity. If future studies confirm the connection between lymphopenia and ATG toxicity, this finding could become important for clinicians using ATG because they could then adjust ATG to lymphocyte counts. Thus optimal dose and schedule of ATG that are of benefit for GVHD but do not induce overimmunosuppression needs to be addressed. A prospective, multicenter, open-label, randomized phase 3 study of ATG in transplantation from an HLA-identical sibling showed a significantly lower rate of chronic GVHD in the ATG group, whereas overall survival was similar.
Another important approach to reduce GVHD is posttransplant cyclophosphamide (PT-Cy) in haploidentical transplant recipients.5 The broad application of this approach to patients with leukemia not in complete remission may incur an increased risk for relapse based on preclinical graft-versus-leukemia studies showing a loss of the donor polyclonal T-cell pool and graft-versus-leukemia effects when PT-Cy was given.6
Cellular therapy approaches against acute GVHD including T regulatory cells (Tregs) and mesenchymal stroma cells are not discussed here because of length restrictions but can be found in previous reviews.1
Targeting cell-surface molecules including cytokine receptors, chemokine receptors, integrins, and costimulatory molecules in GVHD
Different pro-inflammatory cytokines have been shown to be functionally involved in acute and chronic GVHD (reviewed in Zeiser and Blazar1,2 ). These cytokines include interleukin-1β (IL-1β), IL-2, IL-6, IL-11, IL-12, IL-15, IL-17, IL-18, IL-21, IL-23, IL-33, interferon-γ, and tumor necrosis factor-α (reviewed in Zeiser and Blazar1 ). The initial release of many of these cytokines is not GVHD related but the result of tissue damage from cytotoxic conditioning or infections. The resulting cell death leads to a shift of intracellular molecules into the extracellular space. Although these molecules do not activate the immune system when they are retained intracellularly, their occurrence in the extracellular space can provoke strong immune responses (reviewed in Zeiser et al7 ). Molecules that can act as danger-associated molecular patterns (DAMPs) include adenosinetriphosphate,8 hyaluronic acid, HMGB-1, S100 protein,9 and uric acid,10 among others. Besides these, the cytokine production is caused by a pathogen-associated molecular pattern (PAMP) such as lipopolysaccharides. Activation of surface receptors and intracellular sensors of DAMP and PAMP provoke intracellular biochemical cascades that cause cytokine transcription and cleavage of inactive cytokines stored intracellularly. An example for such a process is activation of the Nlrp3 inflammasome by the DAMP adenosinetriphosphate and uric acid, which leads to pro-IL-1β cleavage into its bioactive form that is consecutively secreted and promotes GVHD. Besides the amplification of proinflammatory signals, some cytokines cause direct cytotoxicity on GVHD target cells, such as tumor necrosis factor-α, that induced apoptosis in epithelial cells. The role of cytokines is often time- and tissue-context dependent. One example is IL-33, which has anti-inflammatory properties, given before tissue damage based on the expansion of IL-33 receptor (suppressor of tumorigenicity) expressing Treg.11 In contrast IL-33 administration during GVHD promotes interferon-γ producing T-cell expansion and acute GVHD.11
Different strategies that target cytokines directly or their receptors were tested in clinical studies. Examples are inolimomab and daclizumab, which bind to the IL-2 receptor α chain CD25. A study on daclizumab/infliximab for steroid refractory (SR) GVHD showed a dismal outcome: all patients died a median of 35 days from initiation of therapy.12 This could be due to the loss Treg suppression because IL-2 has not only pro-inflammatory function, but also expands anti-inflammatory Treg.13 The effect on Treg could help to explain why a randomized phase 3 randomized trial showed no advantage of inolimomab compared with ATG in patients with SR-GVHD.14 Also, blockade of IL-1β and IL-11 did not become a therapeutic option for acute GVHD based on lack of efficacy and toxicity, respectively (reviewed in Zeiser et al15 ). Blockade of IL-6 receptor with tocilizumab in GVHD prophylaxis was effective, whereas so far data on SR-GVHD are not available.16
Besides the activation of T cells by cytokines, they migrate to the lymph nodes where priming takes place and from there to the target organs are key steps in GVHD pathophysiology. Consequently, different strategies blocking chemotaxis have been tested in the clinic. Examples are vedolizumab, an antibody directed against α4β7 integrin used for SR-GVHD,17 and maraviroc, a CCR5 inhibitor, which were both applied to prevent GVHD18 in early clinical trials. α4β7 integrin inhibition was connected to high response rates17 but also to severe infections; a small case series reported that all patients died at a median of 32 days posttreatment.19 Comparable to the cytokine receptor blockade, redundant mechanisms may overrule the protective effects of chemokine receptor blockade (eg, CXCR3 signaling was recently identified as a resistance mechanism to CCR5 blockade in GVHD).20 The Novel Approaches for Graft-versus-Host Disease Prevention Compared to Contemporary Controls (CTN 1203) study showed that maraviroc did not induce lower GVHD rates when compared with bortezomib or PT-Cy.21 Also, preclinical total body irradiation (TBI)-based GVHD models had shown that GVHD severity worsened when T cells were derived from CCR5−/− compared with wild-type donors.22 Another important event in GVHD pathophysiology that can be targeted is costimulation of T cells. Costimulatory pathway signaling is required for full activation and survival of disease causing T cells, which makes them attractive cell-surface targets in GVHD. A role for multiple costimulatory molecules has consistently been shown in GVHD, such as CD28, CD40L, OX40, 4-1BB, and ICOS, but also negative regulatory pathways such as CTLA-4, PD-1/PD-L1, PD-L2, B- and T-lymphocyte attenuator, lymphotoxin-like compete with herpes simplex virus glycoprotein D for HVEM, a receptor expressed by T lymphocytes, lymphocyte-activation gene 3 (LAG3), T-cell immunoreceptor with Ig and ITIM domains, and B7-H3 (reviewed in Zeiser et al15 ). One example of costimulatory blockade that has been tested in clinical trials for acute GVHD is abatacept, which is a fusion protein composed of the Fc region of the immunoglobulin IgG1 fused to the extracellular domain of CTLA-4. In a small case series, SR-GVHD patients were treated combining abatacept along with etanercept and basiliximab, yielding an overall response rate of 40% at day 56.23
Overall, these preclinical and clinical studies show that targeting cell surface molecules such as cytokine receptors, chemokine receptors, integrins, and costimulatory molecules have activity against GVHD. A better understanding of GVHD-resistant mechanisms, redundant pathways, and timing of therapy will be important to improve the outcome of patients treated with these approaches.
Signaling pathways as therapeutic targets in GVHD
Based on the broad array of cytokines and chemokines involved in GVHD pathogenesis, it is conceivable that depletion of 1 cytokine will not prevent disease, because many cytokines have redundant functions. This concept is not novel, but in recent years, small molecules that target signaling molecules have become available because sequencing analyses of cancer tissues have revealed activating oncogenic mutations. Many of the oncogenic mutations occur downstream of signaling pathways that are also central for cytokine or growth factor signaling and the novel inhibitors of oncogenic pathways are often also immunomodulatory. Signaling pathways that were studied in preclinical GVHD models include MEK, Aurora kinase A (AURKA), JAK 1/2, CDK2/5 and phosphatidylinositol-4,5-bisphosphate 3-kinase (Figure 1), besides multiple others (reviewed in Zeiser and Blazar1,24 ).
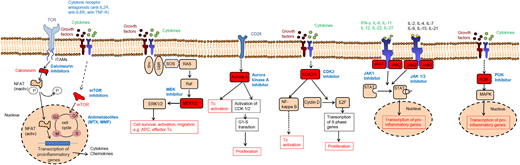
Therapeutic targets in GVHD. The simplified scheme shows the mode of action of multiple immunosuppressive strategies. Although calcineurin inhibitors, mTOR inhibitors, and antimetabolites are already in clinical routine practice, all other approaches are experimental and their effect has been tested mainly in preclinical models or in vitro. Selected kinases that have been subject to targeted therapy approaches in acute GVHD are shown. Blockade of the kinases ROCK-1, Aurora A, CDK2, MEK-1/2, JAK1/2, and PI3K were shown to reduce alloreactive T-cell activation. MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin, MTX, methotrexate.
Therapeutic targets in GVHD. The simplified scheme shows the mode of action of multiple immunosuppressive strategies. Although calcineurin inhibitors, mTOR inhibitors, and antimetabolites are already in clinical routine practice, all other approaches are experimental and their effect has been tested mainly in preclinical models or in vitro. Selected kinases that have been subject to targeted therapy approaches in acute GVHD are shown. Blockade of the kinases ROCK-1, Aurora A, CDK2, MEK-1/2, JAK1/2, and PI3K were shown to reduce alloreactive T-cell activation. MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin, MTX, methotrexate.
A preclinical study reported that the RAS/MEK/extracellular signal-regulated kinase pathway was preferentially activated in naive and central memory human T cells, which are disease-causing T cells in GVHD.25 Consistent with this finding, the authors observed that MEK inhibition preferentially inhibited alloreactive T cells while sparing virus-specific T cells.25 Another kinase inhibited for GVHD treatment in preclinical studies was AURKA.26 Here the rationale was that the gene expression profile of nonhuman primate T cells during acute GVHD indicated AURKA as a druggable target.26 The authors later confirmed this target in mouse GVHD studies showing that GVHD could be reduced by AURKA inhibition.26
The first topical kinase inhibitor approach was recently reported by the group of Teshima.27 The authors found that GVHD affects Lgr5+ hair follicle stem cells, which can be blocked by topical ruxolitinib. This novel strategy was able to protect skin stem cells and maintain skin homeostasis in GVHD.27 JAK1/2 inhibition was also shown to reduce GVHD when given systemically in mice,28 and a retrospective study suggested a benefit in patients with SR-GVHD.29 Because of the retrospective nature of the clinical study, these data have to be interpreted with caution. However a prospective clinical trial is ongoing to clarify if JAK1/2 inhibition can improve the outcome of patients developing SR-GVHD. Targeting JAK1/2 may not only affect T cells but also myeloid cells such as dendritic cells (DCs) and neutrophil granulocytes (neutrophils), which contribute to GVHD. DC were shown to promote GVHD and inhibition of JAK1/2 reduced expression of the transcription factor CIITA, which activates the major histocompatibility complex class II promoter30 ; therefore, the capacity of DC to stimulate the donor T cells may be reduced. Other GVHD promoting myeloid cells are recipient neutrophils that are activated early after allo-HCT in the intestinal tract, in particular in the ileum.31 These activated neutrophils can promote GVHD through their activation and reactive oxygen species production in the intestinal tract.32
In aggregate, the inhibition of different intracellular signaling pathways may become an attractive strategy to prevent or treat GVHD. It may be interesting to simultaneously block different pathways, such an approach combining AURKA and JAK2 was shown to reduce GVHD potently in preclinical studies.33 It is also conceivable that a combination of kinase inhibition with other strategies such as extracorporeal photopheresis 34 will yield higher response rates in SR-GVHD.
Microbiome modification
There is increasing evidence that a reduced diversity of the intestinal microbiome is connected to a higher incidence of GVHD.35 The studies also describe that certain bacteria in the intestinal tract decline upon antibiotic treatment in mice and patients.35,36 These insights are valuable for our understanding of the role of the microbiome in GVHD and indicate to the transplant physicians that it is important to carefully evaluate if broad-spectrum antibiotic treatment is needed. The concept to protect the intestinal microbiome is further supported by studies showing that antibiotic-based bacterial depletion will reduce microbiota-derived metabolites such as butyrate, which is crucial for intestinal homeostasis. Butyrate, secreted by different intestinal bacteria, is a histone deacetylase inhibitor that modulates GVHD in an indoleamine-2,3-dioxygenase–dependent manner.37
In contrast to these beneficial effects, the direct penetration of microbiota through the intestinal wall in an immunodeficient allo-HCT patient is unfavorable. Therefore, from a clinical perspective, patients that develop fever or signs of sepsis need broad-spectrum antibiotics even if this may increase their risk for acute GVHD. Direct effects of bacterial components that activate pattern recognition receptors, such as Toll like receptors and nucleotide-binding oligomerization domain-like receptors (NOD-like receptors) that activate antigen-presenting cells may promote local and inflammation, which triggers GVHD.38 Not only bacteria but also fungi and viruses were studies in the context of GVHD. α-Mannan derived from fungi was shown to cause Th17-mediated pulmonary GVHD in mice.39 A recent study that analyzed the longitudinal intestinal virome in 44 allo-HCT recipients using metagenomics reported an expansion of the overall proportion of vertebrate viral sequences in patients undergoing allo-HCT.40 The authors also reported persistent DNA viruses over time in individuals with enteric GVHD compared with allo-HCT without GVHD.40 The transfer of microbial species into the intestinal tract as a form of acute GVHD therapy has been evaluated. In a small pioneer study, the first successful and safe application of related fecal microbiota transplants (FMTs) via nasoduodenal tubes in patients suffering from SR-acute GVHD was reported.41 The trial was designed to evaluate safety of FMT and, as an observation, the authors report that GVHD improved in 3 of 4 patients 28 days after first FMT.41 More recently, a prospective open-label pilot study reported the results using third-party FMT capsules in patients undergoing allo-HCT.42 FMT capsules were administered after neutrophil engraftment, and antibiotics were not allowed within 48 hours before FMT.42 The authors reported an improvement in intestinal microbiome diversity after FMT that was associated with expansion of stool-donor taxa.42 Although these studies indicate that third-party FMT after allo-HCT appears to be feasible, safe, and can lead to expansion of recipient microbiome diversity, FMT is still to be considered an experimental treatment approach and needs further clinical validation.
Tissue repair approaches against GVHD
Besides strategies that modulate the microbiome, other strategies aim at protecting or regenerating intestinal stem cells (ISCs) and Paneth cells. Examples include IL-22, R-spondin-1 (R-Spo1) and keratinocyte growth factor (KGF) (Table 1).
Tissue repair approaches (alphabetical order)
| Name | Source and function | Role in GVHD | Species analyzed | Reference |
|---|---|---|---|---|
| IL-22 | Produced by ILC3, splenic LTi-like cells, Th17 cells | IL-22 is protective in mouse models of acute GVHD | Mouse | 43,44 |
| Promotes ISC expansion | Clinical trial on IL-22 IgG2-Fc (F-652), NCT02406651 | Human | ||
| Can trigger inflammation in the presence of IL-17S | ||||
| KGF | Produced by intraepithelial TCR γ/δ cells and mesenchymal cells | Reduced acute GVHD in mouse models | Mouse | 46 |
| Leads to expansion of intestinal epithelial cells | A phase 1/2 randomized placebo-controlled trial showed that KGF did not reduce acute GVHD but did lower mucositis rates in patients conditioned with more intensive TBI | Human | 47 | |
| R-spo-1 | Produced by the developing central nervous system, as well as in the adrenal glands, ovary, testis, thyroid, and trachea | R-Spo1 reduced acute GVHD in the mouse model by protecting ISC from conditioning injury | Mouse | 49,50 |
| R-spo1 induces increased internalization of LGR5 on ISC and Paneth cells, which leads to their expansion | R-Spo1 protects Paneth cells and leads to a more diverse intestinal microbiome |
| Name | Source and function | Role in GVHD | Species analyzed | Reference |
|---|---|---|---|---|
| IL-22 | Produced by ILC3, splenic LTi-like cells, Th17 cells | IL-22 is protective in mouse models of acute GVHD | Mouse | 43,44 |
| Promotes ISC expansion | Clinical trial on IL-22 IgG2-Fc (F-652), NCT02406651 | Human | ||
| Can trigger inflammation in the presence of IL-17S | ||||
| KGF | Produced by intraepithelial TCR γ/δ cells and mesenchymal cells | Reduced acute GVHD in mouse models | Mouse | 46 |
| Leads to expansion of intestinal epithelial cells | A phase 1/2 randomized placebo-controlled trial showed that KGF did not reduce acute GVHD but did lower mucositis rates in patients conditioned with more intensive TBI | Human | 47 | |
| R-spo-1 | Produced by the developing central nervous system, as well as in the adrenal glands, ovary, testis, thyroid, and trachea | R-Spo1 reduced acute GVHD in the mouse model by protecting ISC from conditioning injury | Mouse | 49,50 |
| R-spo1 induces increased internalization of LGR5 on ISC and Paneth cells, which leads to their expansion | R-Spo1 protects Paneth cells and leads to a more diverse intestinal microbiome |
TCR, T-cell receptor.
IL-22 enhances the regeneration of ISC that express IL-22 receptors. Elegant studies by the van den Brink and Hanash groups show the critical role of IL-22 produced by innate lymphoid cells type-3 for ISC protection and regeneration.43,44 The authors decipher the signaling events and show that IL-22 induced STAT3 phosphorylation in Lgr5+ ISCs, and STAT3 was crucial for both organoid formation and IL-22–mediated regeneration.44 To test the preclinical findings in patients, a clinical trial on IL-22 IgG2-Fc (F-652) for grade 2-4 lower gastrointestinal GVHD is currently recruiting (NCT02406651). In contrast to acute GVHD, donor T cell–derived IL-22 was reported to promote cutaneous chronic GVHD.45 The pro-inflammatory vs tissue-protective roles of IL-22 seem to be regulated by the simultaneous presence of IL-17A.
Another protein tested for its effect on acute GVHD is KGF, which can be produced by intraepithelial T-cell receptor γ/δ cells and mesenchymal cells. KGF is not selective for ISC, but rather for different intestinal epithelial cells. Exogenous KGF administration, designed to increase epithelial cell numbers before radiation, reduced GVHD in mouse models.46 Based on these findings in mouse models of acute GVHD, KGF (Palifermin, Kepivance) was tested in a phase 1/2 randomized placebo controlled trial.47 KGF did lower mucositis rates in patients conditioned with more intensive TBI vs chemotherapy regimens, but GVHD was not lower in the KGF group.47 The clinical response to KGF is likely to be due to epithelial proliferation and mucosal thickening. A more recent analysis of several studies showed that patient-reported outcome was improved upon KFG administration after cytotoxic treatment.48
A major signaling pathway that promotes intestinal cell growth is canonical WNT/β catenin signaling pathway. LGR5 is a member of the Wnt signaling pathway, which has been shown that costimulation with R-spo1 and Wnt-3a induce increased internalization of LGR5. Treatment with R-Spo1 reduced acute GVHD in the mouse model by protecting ISC.49 Recent studies extend these observations by showing that not only ISC but also Paneth cells expand upon R-Spo1 treatment.50 Paneth cell secretion of antimicrobial peptides is critical to maintain intestinal homeostasis and microbiome stability. In agreement with this, the protective effect of R-Spo1 was partly abrogated when mice were treated with broad-spectrum antibiotics.49
These different tissue-regenerative approaches may become complementary to the immunosuppressive approaches that are widely used to treat GVHD. However, the local cytokine milieu (eg, presence of IL-17A) and the potential to promote malignancies (WNT/β catenin signaling) have to be considered when these approaches are further developed in the clinic.
Summary
Major advances have been made in the understanding of the biology of acute GVHD, which has shaped the portfolio of novel therapy approaches. Although cytokine antagonism has shown heterogenous response patterns, other strategies that inhibit signaling pathways that promote GVHD have been developed from the mouse model into clinical trials. Because immunosuppressive strategies increase the risk of relapse and infections, tissue-regenerative approaches that promote healing of GVHD-related tissue damage and reinstallment of a diverse microbiome may be important to complement the classical GVHD therapy approaches.
Acknowledgments
I apologize to those whose work could not be cited because of space limitations.
Correspondence
Robert Zeiser, Department of Hematology, Oncology and Stem Cell Transplantation, Freiburg University Medical Center, Faculty of Medicine, Albert-Ludwigs University Freiburg, Hugstetter Str 55, 79106 Freiburg, Germany; e-mail: robert.zeiser@uniklinik-freiburg.de.
