
Abstract
Anemia is common in older persons, and often remains unexplained despite a thorough clinical history, physical examination, and focused laboratory testing, including marrow aspiration, biopsy, and karyotyping. The advent of molecular genetic testing panels in hematology clinical practice has complicated the evaluation of older patients with unexplained anemia. While the presence of a somatic mutation provides evidence of clonal hematopoiesis and may support a diagnosis of a hematologic neoplasm such as one of the myelodysplastic syndromes (MDS), with rare exceptions, individual mutations are not strongly associated with one specific diagnosis, nor are they by themselves diagnostic of neoplasia. A clonal mutation in a patient with cytopenias and a nondiagnostic bone marrow may indicate a syndrome with a similar natural history to MDS, but at present there are no clear criteria to distinguish cytopenias coincidentally seen in association with an unrelated clonal mutation from cytopenias that are directly caused by that mutation. Ongoing and planned analyses will help define when mutation patterns alone can identify a disorder equivalent to a morphologically defined myeloid neoplasm such as MDS, further clarifying the etiology and natural history of unexplained anemia in the elderly.
Learning Objectives
Describe the common causes of anemia in older persons and recommend appropriate diagnostic testing
Evaluate the results of molecular genetic testing of patients with unexplained anemia or other cytopenias in the context of emerging data on clonal hematopoiesis in healthy persons
Distinguish clonal hematopoiesis of indeterminate potential, clonal cytopenias of undetermined significance, idiopathic cytopenias of undetermined significance, and MDS
Introduction
Hematologists are all too aware that anemia is exceptionally common in the general population, especially in older persons.1,2 Anemia is associated with a broad range of potential causes, and establishing a specific etiology for anemia is a critical task for physicians evaluating patients, because many causes of low hemoglobin are reversible and treatment may prevent adverse outcomes.
However, despite appropriate diagnostic testing and a careful search for common causes of anemia in adults (eg, occult bleeding, nutritional deficiency, inflammation, deficient erythropoietin synthesis, late presentation of a congenital disorder of hemoglobin synthesis, a marrow failure syndrome, drug or toxin effect, hemolysis, or a hematologic neoplasm), anemia often remains unexplained, especially mild anemia in the elderly.3 What has made evaluation of unexplained anemia and other cytopenias more complex in recent years is the progressive introduction into clinical practice of molecular genetic testing panels targeted to genes involved in the pathobiology of myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), myeloproliferative neoplasms (MPN), and other hematological cancers.4,5 High-throughput, massively parallel DNA sequencing–based assays are powerful tools that are now routinely ordered by clinicians searching for a cause of unexplained cytopenias in their patients, but the results need to be interpreted with caution.
Though the cost billed to patients and their insurers for these molecular genetic tests varies widely based on nontechnical factors such as negotiated contractual pricing, from the standpoint of consumable reagents, amortized durable equipment and technician time, next-generation sequencing assays are now often less expensive than traditional hematopathology tests such as marrow biopsy with immunohistochemistry and professional morphologist interpretation. In addition, molecular assay results are available much more quickly than conventional G-banded metaphase karyotyping. At the author’s institution, for instance, a 98-gene panel assay (Table 1) is performed on all new patients with known or suspected MDS, MPN, or leukemia; results with basic interpretation are provided to clinicians in 72 to 96 hours, and the cost is less than that for single-gene Sanger sequencing in an external reference laboratory.6 The advent of this assay with provision of results in “real time” has greatly influenced patient care, and discussion of the biological meaning and clinical implications of assay results are the subject of many clinicopathologic conferences.
Targeted genes/exons in a representative molecular genetic testing panel focused on hematologic malignancies
| ABL1 e2-e10 | ASXL1 e1-e13 | ATM e2-e63 | BCL11B e4 |
| BCOR e2-e15 | BCORL1 e1-e12 | BRAF e15 | BRCC3 e3-e11 |
| CALR e9 | CBL e7-e8 | CBLB e9-e11 | CD79B e5-e6 |
| CEBPA e1 | CNOT3 e1-e2 | CREBBP e2-e21, e23-31 | CRLF2 e6 |
| CSF1R e22 | CSF3R e14-e18 | CTCF e3-e12 | CTNNB1 e2-e4 |
| CUX1 e1-e21 | CXCR4 e2 | DNMT3A e2-e23 | DNMT3B e2-e23 |
| EED e1-e12 | EGFR e18-e21 | EP300 e18-e27 | ETV6 e1-e8 |
| FANCL e1-e14 | FBXW7 e8-e12 | EZH2 e2-e8, e11-e20 | FLT3 e14-e16, e20 |
| GATA1 e2-e6 | GATA2 e2-e6 | GATA3 e4-e6 | GNAS e8-e9 |
| GNB1 e5-e6 | IDH1 e4 | IDH2 e4 | IKZF1 e2-e8 |
| IKZF2 e1-e8 | IKZF3 e1-e8 | IL7R e6 | JAK1 e10-e25 |
| JAK2 e12, e14 | JAK3 e11-e24 | KIT e8-9, e11, e17 | KRAS e2-e5 |
| LUC7L2 e3-e11 | MAP2K1 e2-e3 | MEF2B e3 | MPL e10 |
| MYD88 e5 | NOTCH1 e24-e28 | NOTCH1 e34 | NOTCH2 e24-e28 |
| NOTCH2 e34 | NOTCH3 e25-e26 | NOTCH3 e33 | NPM1 e10-e11 |
| NRAS e2-e5 | PAX5 e3, e6-e7 | NT5C2 e9, e11, e13, e15, e17 | |
| PDS5B e3-e35 | PHF6 e2-e10 | PDGFRA e10-e21, e23 | PIGA e2-e6 |
| PIM1 e1-e6 | PRPF40B e2-e26 | PIK3CA e2, e10, e21 | PRPF8 e2-e43 |
| PTEN e1-e9 | PTPN11 e1-e15 | RAD21 e2-e14 | RET e7 |
| RIT1 e1-e6 | RPL10 e5 | RUNX1 e2-e9 | SETBP1 e4 |
| SF3B1 e12-e16 | SF1 e1-e10, e13 | SF3A1 e1-e2, e5-e16 | SETD2 e1-e4, e6-e21 |
| SH2B3 e2-e8 | SMC1A e1-e25 | SMC3 e2-e29 | SRSF2 e1 |
| STAG2 e3-e35 | TET2 e3-e11 | STAT3 e2-e17, e21-23 | TLR2 e1 |
| TP53 e2-e11 | U2AF1 e2, e6 | U2AF2 e1-e12 | WHSC1 e17-e18 |
| WT1 e1-e10 | XPO1 e15-e16 | ZRSR2 e1-e11 |
| ABL1 e2-e10 | ASXL1 e1-e13 | ATM e2-e63 | BCL11B e4 |
| BCOR e2-e15 | BCORL1 e1-e12 | BRAF e15 | BRCC3 e3-e11 |
| CALR e9 | CBL e7-e8 | CBLB e9-e11 | CD79B e5-e6 |
| CEBPA e1 | CNOT3 e1-e2 | CREBBP e2-e21, e23-31 | CRLF2 e6 |
| CSF1R e22 | CSF3R e14-e18 | CTCF e3-e12 | CTNNB1 e2-e4 |
| CUX1 e1-e21 | CXCR4 e2 | DNMT3A e2-e23 | DNMT3B e2-e23 |
| EED e1-e12 | EGFR e18-e21 | EP300 e18-e27 | ETV6 e1-e8 |
| FANCL e1-e14 | FBXW7 e8-e12 | EZH2 e2-e8, e11-e20 | FLT3 e14-e16, e20 |
| GATA1 e2-e6 | GATA2 e2-e6 | GATA3 e4-e6 | GNAS e8-e9 |
| GNB1 e5-e6 | IDH1 e4 | IDH2 e4 | IKZF1 e2-e8 |
| IKZF2 e1-e8 | IKZF3 e1-e8 | IL7R e6 | JAK1 e10-e25 |
| JAK2 e12, e14 | JAK3 e11-e24 | KIT e8-9, e11, e17 | KRAS e2-e5 |
| LUC7L2 e3-e11 | MAP2K1 e2-e3 | MEF2B e3 | MPL e10 |
| MYD88 e5 | NOTCH1 e24-e28 | NOTCH1 e34 | NOTCH2 e24-e28 |
| NOTCH2 e34 | NOTCH3 e25-e26 | NOTCH3 e33 | NPM1 e10-e11 |
| NRAS e2-e5 | PAX5 e3, e6-e7 | NT5C2 e9, e11, e13, e15, e17 | |
| PDS5B e3-e35 | PHF6 e2-e10 | PDGFRA e10-e21, e23 | PIGA e2-e6 |
| PIM1 e1-e6 | PRPF40B e2-e26 | PIK3CA e2, e10, e21 | PRPF8 e2-e43 |
| PTEN e1-e9 | PTPN11 e1-e15 | RAD21 e2-e14 | RET e7 |
| RIT1 e1-e6 | RPL10 e5 | RUNX1 e2-e9 | SETBP1 e4 |
| SF3B1 e12-e16 | SF1 e1-e10, e13 | SF3A1 e1-e2, e5-e16 | SETD2 e1-e4, e6-e21 |
| SH2B3 e2-e8 | SMC1A e1-e25 | SMC3 e2-e29 | SRSF2 e1 |
| STAG2 e3-e35 | TET2 e3-e11 | STAT3 e2-e17, e21-23 | TLR2 e1 |
| TP53 e2-e11 | U2AF1 e2, e6 | U2AF2 e1-e12 | WHSC1 e17-e18 |
| WT1 e1-e10 | XPO1 e15-e16 | ZRSR2 e1-e11 |
List of 98 genes and exons included in the “Rapid Hematology Panel” test at Dana-Farber Cancer Institute/Brigham & Women’s Hospital as of May 1, 2016. The majority of genes recurrently mutated in myeloid or lymphoid neoplasms are included in this list. Gene/exon list developed by Jon C. Aster, Frank C. Kuo, and Neal I. Lindeman (Brigham & Women’s Hospital Center for Advanced Molecular Diagnostics), and R. Coleman Lindsley (Dana-Farber Cancer Institute Division of Hematological Malignancies) in consultation with clinical hematology groups.
Confounding the simple interpretation of such panels is the recent discovery that more than 10% of the general population over age 70 years carries mutations in genes associated with myeloid neoplasms, usually single mutations at a low variant allele frequency (VAF), defined as the proportion of sequencing reads for a specific gene segment that exhibit a mutation, and proportional to clone size when corrected for copy number.7,8 Patients with MDS or other myeloid neoplasms, in contrast, often have more than one mutation, and these mutations may be present at higher VAF (>20% of sequencing reads).9 Because >90% of patients with MDS have a detectable somatic mutation in one of about 25 recurrently mutated genes (Figure 1),10 a negative result on a molecular genetic panel that includes all of these commonly mutated genes has a high negative predictive value for MDS.11 However, if a patient has cytopenias and a clonal mutation, we currently do not know how to distinguish cytopenias caused by a nonclonal cause that are coincidentally present together with an unrelated clonally restricted mutation from those cytopenias that are pathophysiologically related to a clonal process. This is a major limitation that will need to be addressed in future studies.

“Word cloud” of mutations recurrently associated with MDS. Many of these mutations are commonly included on currently available molecular genetic testing assays (“mutation panels”) designed to aid in diagnosis and prognosis of patients with myeloid neoplasms. Gene font size is proportionate to the square root of mutation frequency, to allow readability of infrequent recurrent mutations, and color corresponds with putative or known biological function of encoded proteins. Data on mutation frequency from Haferlach.10 Figure cocreated with Rafael Bejar, University of California–San Diego.
“Word cloud” of mutations recurrently associated with MDS. Many of these mutations are commonly included on currently available molecular genetic testing assays (“mutation panels”) designed to aid in diagnosis and prognosis of patients with myeloid neoplasms. Gene font size is proportionate to the square root of mutation frequency, to allow readability of infrequent recurrent mutations, and color corresponds with putative or known biological function of encoded proteins. Data on mutation frequency from Haferlach.10 Figure cocreated with Rafael Bejar, University of California–San Diego.
Epidemiology
The most widely cited data on the prevalence of anemia in the United States come from the Third National Health and Nutrition Examination Survey (NHANES III), conducted from 1988 to 1994.12 In the NHANES III data set (>33 000 persons aged 2 or older), 11.0% of men and 10.2% of women 65 years and older were anemic, whereas >20% of people older than 85 years were anemic, and anemia was unexplained after basic testing in 34%.12 In other published series, 24% of hospitalized patients ≥65 years13 and 48% to 59.6% of residents of skilled nursing facilities14,15 were noted to have anemia; in many of these patients as well, anemia remained unexplained despite initial evaluation.
The prevalence of anemia depends on the specific hemoglobin value used to define anemia. The 1968 World Health Organization (WHO) thresholds of hemoglobin <13.0 g/dL for men and <12.0 g/dL for nonpregnant adult women are widely used in epidemiologic studies, but these thresholds are usually different from local laboratory normal ranges; they have been criticized and alternate proposals have been put forward.16 “Normal” hemoglobin is population specific: the 95% population range of values defined as normal is lower in people of African descent, which is only partially accounted for by thalassemia alleles and a higher prevalence of iron deficiency, whereas the normal hemoglobin range is higher in people living at high altitude.17
Even mild degrees of anemia are associated with excess morbidity and mortality.18,19 In part this is likely the result of health effects of the underlying cause of anemia, but anemia itself appears to be a risk factor for or is associated with many complications, including geriatric health problems such as frailty, cognitive dysfunction, and falls.19 In some longitudinal assessments of patients with specific conditions such as cardiovascular disease, mortality is increased not only in anemic patients but is also higher for patients with hemoglobin levels in the lowest quartile that extends into the “normal” range, compared with patients with higher hemoglobin levels.20
Pathophysiology of anemia in the elderly
Many different conditions can contribute to anemia, as outlined here before, and an orderly approach is necessary to evaluate patients efficiently and cost-effectively.21 Differential diagnoses can be formulated based on the mean cell volume, a review of the peripheral smear in conjunction with the reticulocyte count, or consideration of the anemia from a pathophysiologic standpoint, because there are only 3 basic mechanisms of anemia: blood loss from the circulation, hemolysis, and erythropoietic failure.
The 3 most common causes of anemia in older persons include nutritional deficiency (especially iron, vitamin B12, and folate); anemia of inflammation; and erythropoietin (EPO) deficiency caused by kidney disease. The diagnostic performance of specific tests for these maladies is beyond the scope of this review but not always straightforward; for instance, “normal” levels of serum B12 may be present despite deficiency, and the serum EPO level must be interpreted in the context of the degree of appropriate response for a given hemoglobin level, rather than the laboratory normal range for EPO. Newer tests can help, such as soluble transferrin receptor, which can evaluate for iron deficiency in the presence of inflammation that alters results of serum ferritin and serum iron/transferrin saturation.
Although the anemia of inflammation appears to be driven primarily by hepcidin, a master regulator of iron availability to the developing erythron, assays for hepcidin, although obtainable in reference laboratories, are not yet in routine use in clinical practice. Instead, inflammation is often inferred from the clinical history, presence of normocytic or microcytic erythrocyte indices, or other markers.22 Inflammatory markers such as C-reactive protein may be elevated even in the absence of a clear cause such as a connective tissue disease or culturable/serologically detectable infection.3,23 Conversely, “anemia of chronic disease” may erroneously be attributed to any chronic condition, including hypertension, hypothyroidism, or diabetes mellitus—disorders that lack a major inflammatory component and are not by themselves causes of anemia.
The degree of renal dysfunction that leads to anemia is variable. Although EPO levels tend to drop to values that can contribute to anemia when the estimated glomerular filtration rate (EGFR) falls below 40 mL/min, some patients continue to make adequate levels of EPO at lower EGFR levels, whereas some patients will also have a blunted and inadequate EPO response to anemia at a higher EGFR, a result of disproportionate dysfunction of the endocrine function of the kidney compared with the filtration function.24
Efforts to explain “unexplained” anemia
One of the major clinical concerns with respect to unexplained anemia in elderly persons is the possibility of MDS. In the NHANES III data set, 17% of patients had features suggestive of MDS or another myeloid neoplasm, such as an elevated mean corpuscular volume or another cytopenia.12 In an Israeli geriatric hospital, 15% of cognitively impaired hospitalized patients with unexplained cytopenias at the time of admission were found on subsequent evaluation to have evidence of MDS.25
Although MDS is important to diagnose because the natural history of MDS includes a risk of death from progressive cytopenias or evolution to AML, securing a diagnosis of MDS may be challenging, particularly in early stages of disease when disease-associated blood and marrow cell morphology changes can be subtle. Current WHO diagnostic criteria for MDS require the presence of one or more cytopenias and either 10% dysplastic cells in one or more hematopoietic lineages, 5% to 19% marrow myeloid blast cells, or a recurrent MDS-associated clonal chromosomal abnormality.26-28 A few karyotypic changes that lack diagnostic specificity (ie, -Y, +8, del(20q)) are not sufficient by themselves to diagnose MDS.28 Although a diagnosis of MDS using WHO guidelines requires subjective assessment of the degree of hematopoietic cell dysplasia, dysplasia assessment has a low interoperator reproducibility.29,30 Healthy older persons with normal blood counts often have dysplastic blood and marrow cells similar to those found in patients with MDS.31,32
Other mechanisms for unexplained anemia in the elderly have been proposed. Low testosterone levels may contribute to mild anemia in older men. Some investigators, perhaps influenced by the observation that marrow cellularity progressively declines with age, have proposed progressive stem cell dysfunction and reduction of hematopoietic potential as another common mechanism for senile cytopenias. Although older individuals do have numerous changes in the phenotype and renewal potential of hematopoietic stem cells (HSCs), there is as yet scant evidence to link these changes to unexplained cytopenias in the absence of a clonal process. In murine models, HSCs appear to increase in number with aging, though they display immunophenotypic changes and a decreased ability to generate hematopoietic cells.33 Acquired resistance of developing erythroid cells to EPO and other cytokine signaling is another proposed mechanism for which there is scant clinical evidence.
A clonal cell population in the absence of features consistent with MDS or another WHO-diagnosable hematologic neoplasm could still contribute to cytopenias. For instance, despite the demonstrated ability of such clones to outcompete normal hematopoietic cells in at least a subset of the marrow microenvironment (a necessity for their survival and detection), a mutant clone may still be ineffective at generating mature, differentiated blood cells. Intramedullary apoptosis, which has been invoked as a common mechanism for cytopenias in the presence of the typical hypercellular MDS marrow, and for which there is some biochemical evidence such as aberrant caspase activation, may remove more differentiated cell progeny deriving from mutant clones, and therefore contribute to cytopenias.34 Neoantigens resulting from clonal mutations in genes encoding splicing factors or epigenetic modifiers might trigger an immune response that suppresses further expansion of clonal cells or shortens the lives of their progeny.
Alphabet soup: ICUS, CHIP, CCUS, MDS
Several terms have been used to describe individuals with unexplained cytopenias, clonal mutations, or both (Table 2; Figure 2A).
Definition of clonal and cytopenic states and their relationship to MDS
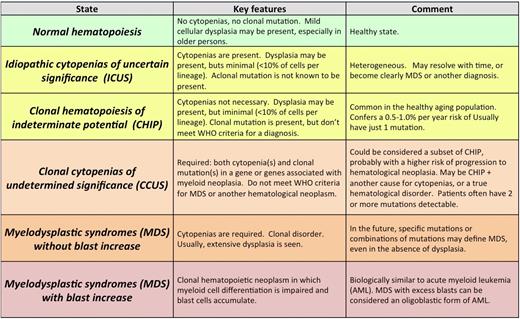
Color represents overall risk to the patient cohort: red and dark orange are higher risk, yellow is lower risk, and green is best. Reprinted from Steensma47 with permission.

Relationship between unexplained cytopenias and mutations. (A) Multiple mutations at high variant allele frequency indicate a clone of substantial size and suggest a high likelihood of a myeloid neoplasm, whereas single mutations at low variant allele frequency is more likely to be CHIP, but the threshold between these states is currently unclear. (B) Venn diagram of the intersection of morphologic dysplasia, clonal mutations and peripheral blood cytopenias. In the future, some patients with cytopenias plus clonal mutations, currently designated as CCUS, may be redefined as MDS. Patients with dysplasia and cytopenias without mutations, in contrast, although currently defined as MDS, may be found to have a distinct syndrome. Such patients appear to be overrepresented in series of responders to immunosuppressive therapy, for example. IDUS, idiopathic dysplasia of undetermined significance.
Relationship between unexplained cytopenias and mutations. (A) Multiple mutations at high variant allele frequency indicate a clone of substantial size and suggest a high likelihood of a myeloid neoplasm, whereas single mutations at low variant allele frequency is more likely to be CHIP, but the threshold between these states is currently unclear. (B) Venn diagram of the intersection of morphologic dysplasia, clonal mutations and peripheral blood cytopenias. In the future, some patients with cytopenias plus clonal mutations, currently designated as CCUS, may be redefined as MDS. Patients with dysplasia and cytopenias without mutations, in contrast, although currently defined as MDS, may be found to have a distinct syndrome. Such patients appear to be overrepresented in series of responders to immunosuppressive therapy, for example. IDUS, idiopathic dysplasia of undetermined significance.
The term idiopathic cytopenia(s) of undetermined significance (ICUS) has been used to describe patients who have a blood cytopenia that remains unexplained despite comprehensive evaluation including marrow examination.35 By definition, patients with ICUS are not known to have a clonal disorder, either because clonality testing has not been performed or results of clonality testing were unrevealing. ICUS is not a specific disorder, but instead a general term that can be used to describe patients in whom cytopenias are present but the specific diagnosis is as yet unclear.26,36 Idiopathic dysplasia of undetermined significance (IDUS) is a more rarely used term to describe the finding of abnormal cell morphology of unclear cause in the absence of cytopenias.37
Although the natural history of ICUS is unclear, there is a risk of subsequent neoplasia diagnosis. In a Mayo Clinic series of 2899 patients evaluated for cytopenias with a bone marrow aspirate and biopsy over a 13-year period, 579 (20%) did not meet criteria for a WHO diagnosis: 182 patients had a morphologically normal marrow, and 397 had a marrow with slightly abnormal morphology that was considered insufficient to diagnose MDS or another disorder.38 Follow-up data were limited in this series, but MDS or AML developed in a few patients, whereas the cytopenias resolved in others; more than one half were found to have one of a wide range of alternate diagnoses, including liver cirrhosis, connective tissue disorders, and low-grade lymphoproliferative neoplasms.
The regional hematology service in Leeds recently reported mutational analysis of 69 patients (of 4875 marrow aspirates obtained to evaluate cytopenias) who had previously had a nondiagnostic marrow biopsy and were subsequently diagnosed with MDS or AML.9 Among these 69 patients, 91% had an MDS-associated mutation detectable by targeted sequencing with a 26-gene MiSeq panel. The clonal mutations detected in this series had significantly greater median VAF compared with the typical VAF of clonal hematopoiesis in healthy people (ie, 40% vs <10%), and multiple mutations also occurred more commonly (≥2 mutations, 64% vs 8% in CHIP series described next). This suggests that there is a higher risk of hematologic malignancy if multiple mutations are present, especially at high VAF (Figure 2B).
A reference diagnostic laboratory in the US compared 249 patients with ICUS to 91 patients with WHO-defined lower-risk MDS with several tools including traditional hematopathology techniques, co-karyotyping, and a 22-gene mutation panel that included MDS-associated genes.11 More than 90% of patients with MDS had a karyotypic abnormality or point mutation, but 40% of those with ICUS also had mutations (and therefore represented CCUS, defined later). Mutation patterns and frequency were similar in the MDS and ICUS series, except that SF3B1, which is strongly associated with the presence of ring sideroblasts (a finding that may give morphologists more confidence in diagnosing MDS),39 was overrepresented in MDS compared with ICUS, as was U2AF1, whereas another splicing factor, SRSF2, was more common in ICUS for unexplained reasons. TET2, DNMT3A, and ASXL1 represented the most commonly observed mutations in both ICUS and MDS cohorts.
Building on observations that somatic chromosomal mosaicism increases in frequency with age40 and that older women with acquired skewing of X chromosome inactivation patterns sometimes have acquired TET2 mutations,41 sequencing data from large genome-wide association studies of nonhematologic conditions were reviewed to explore the frequency of hematologic neoplasia-associated mutations in people without a known hematologic phenotype, who usually had normal complete blood counts and normal or slightly increased red cell indices.7,8,42 As mentioned before, ∼10% of the population at age 70 have a hematologic malignancy-associated mutation, and the frequency of mutations continues to increase with age.7,8,42
We have proposed the term clonal hematopoiesis of indeterminate potential (CHIP) to describe these individuals; “indeterminate potential” refers to the statistical probability that these clonal states will evolve to a frank neoplasm, a risk estimated at 0.5% to 1% per year.43 CHIP can be thought of as similar to monoclonal B-cell lymphocytosis or monoclonal gammopathy of undetermined significance, premalignant clonal states that also have a risk of progression to frank neoplasia of similar magnitude to CHIP (∼1% per year).44,45 CHIP has also been termed age-related clonal hematopoiesis, though it can rarely occur in younger patients.7 Notably, CHIP is associated with both an increased risk of hematologic neoplasia, especially when the VAF is high (median 25% in patients who went on to develop a hematologic neoplasm compared with <10% in those who did not), but also with higher all-cause mortality, including cardiovascular disease, for unclear reasons that are the topic of several ongoing investigations.
CHIP is sometimes discovered during staging studies for an unrelated malignancy, but in clinical practice, testing for mutations associated with hematologic neoplasia is usually done as part of the evaluation of unexplained cytopenias.4 CCUS has been used to describe individuals with meaningful unexplained cytopenias (ICUS) who are also found to have a clonal mutation, but who do not meet WHO criteria for MDS or another hematologic malignancy.43,46 Not surprisingly, it is more likely that a mutation will be found in a cytopenic patient than in the general population.7,11,47
Some clinicians might assess patients with CCUS as having morphologically unapparent MDS or another hematologic neoplasm. However, the frequency of CHIP (and of reactive states such as nutritional deficiency or inflammation) compared with MDS suggest that caution is indicated, because the clonal mutation may not actually be responsible for the cytopenias. It seems probable that in the future, specific variant alleles and VAF patterns will define patients with a syndrome like MDS, even in the absence of dysplasia, just as certain MDS-associated karyotypes do so today.
Recommendations
Despite the uncertainty about interpretation in some situations, clinical testing for mutations is reasonable in cases of unexplained anemia, especially if other cytopenias are present. Negative results on a well-designed mutation panel, although not entirely excluding MDS, are reassuring. Sometimes a finding such as a STAT3 mutation (strongly associated with T-cell large granular lymphocyte disorder,48 a diagnosis that is easily missed on routine morphology) or a BRAF mutation (present in virtually all cases of classic hairy cell leukemia49 ) may suggest a specific, treatable diagnosis.
In contrast, a single mutation in one of the common CHIP-associated genes (eg, TET2, DNMT3A) at low VAF (<10%, adjusted for copy number) should be viewed with a degree of skepticism as a cause of cytopenias and other etiologies sought. It is important to follow patients with unexplained cytopenias over time and track evolution, though insurance companies are reluctant to pay for serial molecular genetic assays to assess stability of a clone.
Many panels include mutations that can be germline (eg, GATA2, RUNXI1, ETV6, TP53, Fanconi anemia–associated genes—all of which predispose to myeloid neoplasms50 ), and finding such mutations may have familial implications. Therefore, clinicians should be prepared to have discussions about germline mutations and refer patients to genetic counselors as appropriate.
What is most needed are good natural history studies of ICUS, CHIP, and CCUS populations to more clearly define populations at high risk of disease progression. Only when such results are available will we be able to counsel patients effectively and rationally formulate trials to help prevent clonal progression.
Correspondence
David P. Steensma, Dana-Farber Cancer Institute, Division of Hematological Malignancies, 450 Brookline Ave, Boston MA 02215; e-mail: david_steensma@dfci.harvard.edu.
