
Abstract
Rosai–Dorfman disease (RDD), juvenile xanthogranuloma (JXG), and Erdheim–Chester disease (ECD) are non-Langerhans cell (non-LCH) disorders arising from either a dendritic or a macrophage cell. RDD is a benign disorder that presents with massive lymphadenopathy, but can have extranodal involvement. In most cases, RDD is self-limited and observation is the standard approach. Treatment is restricted to patients with life-threatening, multiple-relapsing, or autoimmune-associated disease. JXG is a pediatric histiocytosis characterized by xanthomatous skin lesions that usually resolve spontaneously. In a minority of cases, systemic disease can occur and can be life threatening. Juvenile myelomonocytic leukemia (JMML), as well as germline mutations in NF1 and NF2, have been reported in children with JXG. Recent whole-exome sequencing of JXG cases did not show the BRAF-V600E mutation, although 1 patient had PI3KCD mutation. ECD is an adult histiocytosis characterized by symmetrical long bone involvement, cardiovascular infiltration, a hairy kidney, and retroperitoneal fibrosis. Central nervous system involvement is a poor prognostic factor. Interferon-α is the standard as front-line therapy, although cladribine and anakinra can be effective in a few refractory cases. More than one-half of ECD patients carry the BRAF-V600E mutation. Currently, >40 patients worldwide with multisystemic, refractory BRAF-V600E+ ECD have been treated with vemurafenib, a BRAF inhibitor, which was found to be highly effective. Other recurrent mutations of the MAP kinase and PI3K pathways have been described in ECD. These discoveries may redefine ECD, JXG, and LCH as inflammatory myeloid neoplasms, which may lead to new targeted therapies.
Learning Objectives
To discuss the clinicopathologic features and treatment strategies of Rosai–Dorfman disease, juvenile xanthogranuloma, and Erdheim–Chester disease, with a particular focus on targeted therapy with BRAF inhibitors
To present recently published genomic findings that support the notion that ECD, like LCH, is a myeloid neoplasm
Uncommon histiocytic disorders, or non-Langerhans cell histiocytoses (non-LCH), are a diverse group of disorders defined by the accumulation of histiocytes that do not meet the criteria for the diagnosis of LCH or hemophagocytic lymphohistiocytosis (HLH).1 The normal human dermis contains immunostimulatory myeloid CD45+ HLA-DR+ dendritic cells (DCs) identified by CD11c and CD1c, as well as an additional population of poorly stimulatory macrophages that are positive for CD163, FXIIIa, and CD14, and negative for CD1a.2 The non-LCH disorders are thought to arise from either a DC or a macrophage cell line,3 and can be divided clinically into 3 major groups : those that are predominantly cutaneous, those that are cutaneous but with a major systemic component and those that are predominantly systemic, although skin involvement may be part of the disease. This chapter will focus on 3 uncommon histiocytic disorders, such as Rosai–Dorfman disease (RDD), juvenile xanthogranuloma (JXG), and Erdheim–Chester disease (ECD).
Rosai–Dorfman disease
Rosai–Dorfman disease (RDD) is a rare histiocytic disorder characterized by a benign proliferation of S100–positive histiocytic cells within the sinus of the lymph nodes and the lymphatic vessels of internal organs.4,5 This disorder was first described in 1965 by Dr Destombes, who reported 2 cases with lymphadenopathy and sinus histiocytosis.6 RDD was subsequently identified as a unique clinicopathologic entity in 2 publications dealing with four and 34 patients, respectively, by Rosai and Dorfman, in 1969 and 1972, under the name “sinus histiocytosis with massive lymphadenopathy” (SHML).7,8 RDD has been reported in >1400 publications on MEDLINE, but most of these articles report only a limited number of cases. In 1990, Rosai and Dorfman published the results of an international registry which included 423 distinct cases, and which remains the main source of information about RDD >20 years later.4
Epidemiology
RDD is most frequently seen in children and young adults, but can occur at any age. Patients presenting with isolated intracranial disease tend to be older. The disease is more common in males and in individuals of African descent. RDD has been reported following bone marrow transplant for precursor-B acute lymphoblastic leukemia, concurrently with Hodgkin's and non-Hodgkin's lymphoma or with other histiocytic disorders, such as LCH and histiocytic sarcoma.
Pathogenesis
The etiology of RDD is unknown. A cytokine-mediated migration of monocytes may be involved in histiocytes accumulation and activation. This functional activation could be triggered by different stimuli, due to the coexistence of RDD and autoimmune diseases, hematological malignancies and post-infectious conditions.9 However, there is no evidence for a viral etiology in RDD. Systemic RDD-like histopathology has been described in 41% of patients with autoimmune lymphoproliferative syndrome (ALPS) type Ia, with TNFRSF6 heterozygous germline mutations affecting the gene encoding Fas,10 and in patients with autoimmune hemolytic anemia (AIHA), systemic lupus erythematous (SLE) and juvenile idiopathic arthritis (JIA).9 Similarly, hereditary syndromes such as Faisalabad Histiocytosis11 and “H” syndrome12 have been reported to have pathologic findings compatible with RDD. Because of the presence of a Rosai–Dorfman pattern in hereditary, autoimmune, and malignant conditions, RDD could be considered today as a pattern rather than a single entity. Based on this notion, we propose that RDD be classified into 5 categories: familial, classical (SHML), extranodal, malignancy-associated, and autoimmune-associated RDD (Table 1).

Diagnosis
RDD diagnosis requires histopathologic examination with immunohistochemistry. Intrasinusal histiocytic proliferation with cells showing emperipolesis (erythrocytes, lymphocytes, and plasma cells are engulfed by histiocytes) are a diagnostic feature of RDD. These histiocytes have a normal activated phenotype. Immunostaining is positive for CD68, CD163, and S100, whereas staining for CD1a is negative. RDD lesions have a moderate expression of IL-6, which could be related to the associated polyclonal plasmacytosis and hypergammaglobulinemia. Table 2 presents the recommended initial diagnostic work-up of patients with RDD.
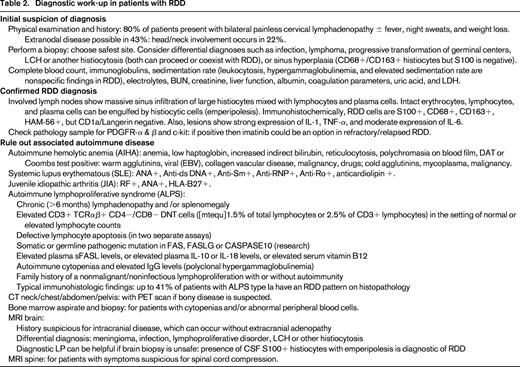
Clinical features
The most frequent clinical presentation of RDD is a massive bilateral and painless cervical lymphadenopathy with fever, night sweats, and weight loss. Mediastinal, inguinal, and retroperitoneal nodes may also be involved. Extranodal involvement by RDD has been documented in 43% of cases with the most frequent sites being skin, soft tissue, upper respiratory tract, multifocal bone, eye and retro-orbital tissue with lymphadenopathy, or as an isolated initial manifestation of disease.4 Other reported sites include urogenital tract, breast, gastrointestinal tract, liver, pancreas, and lungs. Head and neck involvement ha been reported in 22% of cases, most commonly the nasal cavity followed by the parotid gland. Intracranial RDD usually occurs without extracranial lymphadenopathy, and most intracranial lesions are attached to the dura with only few extending intraparynchemally. CNS disease can present clinically and radiographically as meningioma, but the presence of emperipolesis in the CSF is usually diagnostic of RDD.9
Clinical course
The clinical course of RDD is unpredictable with episodes of exacerbation and remissions that could last many years. The disease is often self-limited with a good outcome, nevertheless, 5%-11% of patients die from their disease. Three groups of RDD patients can be identified: (1) patients with only lymph nodes that enlarge suddenly with spontaneous regression and without any further recurrences; these patients can be observed without any treatment; (2) patients with autoimmune disease at presentation who have a more widespread nodal disease and a higher fatality rate; and (3) patients with systemic and multinodal disease, and a protracted clinical course with multiple relapses and remissions for years. In these cases, the severity of disease depends on the type and number of extranodal sites. The latter 2 groups require treatment, however, no standard treatment has been delineated and therapeutic approach is usually determined on a case-by-case basis at a specialist center.
Treatment
Patients with extranodal RDD having vital organ involvement, such as the liver or CNS, or those with nodal disease causing life-threatening complications usually require therapeutic intervention. Surgery is generally limited to biopsy, but debulking may be required in patients with vital organ compromise, such as intracranial dural-based lesions or upper airway obstruction.9 Radiotherapy has limited efficacy in most cases, although it can be beneficial in refractory orbital RDD with visual disturbances.9 Systemic corticosteroids are usually helpful in decreasing nodal size and symptoms, however, they can be immunosuppressive and recurrence of RDD lesions can occur after a short period of interruption.9
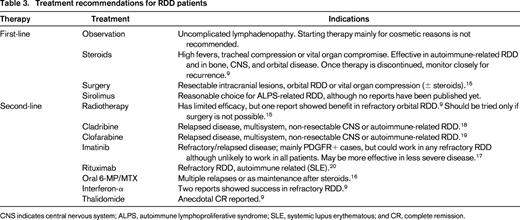
Results with chemotherapeutic agents have not been encouraging. Pulsoni et al reported that only 2 of 10 patients treated with chemotherapy achieved CR, both with combination low-dose methotrexate (MTX) and 6-mercaptopurine (6-MP).15 In another study, a few patients achieved CR after a regimen containing vinblastine, MTX, 6-MP, and 6-thioguanine.16 Anecdotal cases of successful treatment with Interferon-α, acyclovir, and thalidomide have been reported.9 One report described a patient with RDD who showed a rapid and complete response to the tyrosine kinase inhibitor imatinib mesylate. The patients' histiocytes were positive for the imatinib target proteins “platelet-derived growth factor-receptor β” (PDGFRB) and “KIT”. The disease completely responded to treatment with 400-600 mg daily of imatinib for >7 months.17 Drugs that specifically target cytokines (TNF-α and IL-6), such as cladribine or clofarabine, have been found to be effective in recurrent, refractory or severe cases of RDD.18,19 Further, the efficacy of the anti-CD20 monoclonal antibody, rituximab, has been anecdotally described.20 Table 3 illustrates treatment recommendations for patients with RDD.
Juvenile xanthogranuloma
Juvenile xanthogranuloma (JXG), the most common of the non-LCH, is a benign pediatric histiocytosis that usually resolves spontaneously.1 It arises from a dendritic cell, the dermal dendrocyte, which is positive for Factor XIIIa and presumed to be the precursor of many dendritic non-LCH disorders.21 Clinically, JXG involves primarily the skin, but may be localized to a single extracutaneous site without any skin disease, or may be disseminated and life-threatening.
Epidemiology
JXG is a disease of young children (median age 2 years) and may be present at birth. There is a male preponderance, especially in children with multiple skin lesions. Incidence is presumed to be at 1 case/million children,22 but it could be higher because solitary regressing lesions may be missed.
Pathogenesis
The pathogenesis of JXG is unknown, but recent whole-exome sequencing (WES) studies suggest a role of pathologic ERK activation. One study identified 17 somatic mutations by WES in 4 JXG lesions, and although no BRAF-V600E mutations were identified in these lesions, a PI3KCD mutation was identified in 1 patient and a germline NF1 mutation was found in another one with neurofibromatosis type 1 (NF1) and JXG.14 Indeed, JXG has been associated with NF1 and juvenile myelomonocytic leukemia (JMML). In these patients, the JXG usually precedes or occurs concurrently with JMML. Children with JXG and NF1 have 20- to 32-fold increased risk of JMML than patients with NF1 alone.
Diagnosis
JXG diagnosis is confirmed by biopsy to rule out LCH or other benign histiocytoses, dermatofibromas or mastocytosis. Many cases of skin JXG are diagnosed on clinical grounds without histologic confirmation, which is the routine practice when the lesions are typical. Typical histologic features of JXG are giant cells with a ring of nucleus (Touton giant cells), which are also characteristic of ECD. Immunostaining is negative for Langerin, CD1a, and the S100 protein (nevertheless sometimes positive), but classically positive for Factor XIIIa, fascin, CD14, CD68, and CD163. Table 4 shows a classification of the JXG family (personal communication by Dr Ronald Jaffe).

Clinical features
Cutaneous JXG can present as a single (commonest presentation) or multiple papules or nodules, predominantly localized on the face, head and neck, followed by upper torso, upper and lower extremities. During infancy, JXG more commonly presents as multiple, ranging from a few to hundred lesions. Oral JXG can occur as a solitary lesion, without systemic disease and usually at an older age (9 years). Cutaneous JXG usually has a benign course, progressing to yellow-brown lesions followed by gradual involution over months or years.
Systemic JXG occurs in 4% of children with an overall mortality of 5%-10%.23 Median age is usually 3 months and almost one-half of the patients have no skin lesions. The most common presentation is a solitary mass in the deeper soft tissues, followed by liver, spleen, lung, and CNS. CNS involvement with JXG may result in significant complications such as seizures, increased intracranial pressure, diabetes insipidus, developmental delay, and blindness. Radiographically, patients with CNS disease may have leptomeningeal involvement, single, or multiple intracranial or spinal cord lesions. Fatalities due to systemic JXG have been reported mainly due to progressive CNS disease or hepatic failure. Ocular JXG occurs in <1% of children with cutaneous JXG, who are usually very young. Eye involvement is mostly unilateral with red eye and uveitis, glaucoma, spontaneous hyphema, and rarely retina or optic nerve involvement.
Treatment
In general, JXG has an excellent prognosis. For patients with isolated and accessible skin lesions, surgical excision is curative although most childhood lesions will disappear spontaneously.
Most children with JXG require no therapy, but an extensive diagnostic work-up is needed in patients with suspicious systemic disease. Frequent follow-ups in young children (<4 years), particularly those with concurrent NF1, should always include a complete blood count to monitor for JMML. Ophthalmological consultation is recommended for high-risk patients, those <2 years of age, who should undergo screening at diagnosis and every 3 or 6 months until age 2 years. Ocular involvement may require therapy with topical, intralesional, and subconjunctival corticosteroids. Surgery or systemic steroids may be required to treat complications, such as glaucoma or hyphema. Persistent ocular forms with major visual impact can be treated with chemotherapy similar to that used in LCH, such as vinblastine and prednisone. The standard treatment for solitary and symptomatic CNS JXG is surgical resection, provided that surgery is feasible. Patients with unresectable or multifocal cranial JXG have been successfully treated with cladribine24 and vinblastine.25 One pediatric review suggested that symptomatic cases of multisystem JXG, including CNS disease, can successfully be treated with LCH-based regimens.26 Cranial radiation therapy can be considered for unresectable and refractory CNS disease,27 although due to its potentially severe side effects in young children, it is preferable to leave this modality as the last resource. In general, decisions regarding the use of adjuvant therapy require careful attention to potential toxicity in young infants, thus reserving chemotherapy to patients with life-threatening or progressive disease.
Erdheim–Chester Disease
Erdheim–Chester disease (ECD) is a rare non-LCH disorder that was first described as a “lipoid granulomatosis” by Jakob Erdheim and William Chester in 1930.28 ECD, morphologically and immunohistochemically, appears to be a member of the JXG family that involves the long bones in a bilateral fashion.29 ECD can be distinguished from LCH by the characteristic XG immunostaining, which is factor XIIIa+/CD68+/CD163+/fascin+ and S100−/CD1a−/Langerin−/Birbeck granules−.
Epidemiology
Although only 650 ECD cases have been reported up to November 2014, the number has dramatically increased in the last decade due to increased recognition of the disease.30-33 ECD affects predominantly adults between the age of 40 and 70 years (mean age 55 years), and is more frequently diagnosed in males.31 Pediatric cases of ECD have rarely been described (10 cases reported so far), including a 20-month-old child.1
Pathogenesis
The etiology of ECD has been a matter of debate for many years, as it has been considered either as an inflammatory disorder or a clonal neoplastic disease. The RAS-RAF-MEK-ERK pathway is a key cellular signaling pathway that has been found implicated in diverse tumors. Many human tumors carry the BRAF-V600E mutation, causing activation of the RAS-ERK pathway independently of RAS activation. The inhibition of BRAF activation by vemurafenib improves the survival of patients with metastatic BRAF-V600E+ melanomas. In 2010, recurrent somatic activating mutations of the BRAF-V600E were found in 57% of archived LCH lesions.34 Targeted pyrosequencing of paraffin-embedded samples from 127 patients with histiocytoses identified mutually exclusive BRAF-V600E mutations in 54% of ECD samples and 38% of LCH samples, but none of the other non-LCH samples.35 In more recent studies, the frequency of BRAF mutations in ECD has been reported to be between 57% and 75%, depending on the techniques that are used. Therefore, BRAF-V600E appears to be very important in the pathogenesis of ECD, and this is supported by the sustained and reproducible clinical responses in patients with BRAF-V600E+ ECD who are treated with the BRAF inhibitor, vemurafenib.36 Other mutations of the MAP kinase pathway have recently been identified in ECD patients. Indeed, in a large collaborative study between Pitié-Salpêtrière hospital in Paris, France, and the Memorial Sloan Kettering Cancer Center (MSKCC) in New York, PIK3CA and NRAS mutations were found to be recurrent in 11% and 4% of ECD patients with wild-type BRAF-V600E,37 respectively.
Mixed histiocytoses have been described simultaneously in patients at different sites, or in the same lesion or as one disease preceding another; this mixed pattern can occur as LCH/ECD, LCH/JXG, LCH/RDD, or ECD/RDD. In 2014, a multicenter study of 23 patients reported an association between LCH and ECD, linked to the BRAF-V600E mutation.38 ECD either followed LCH (n = 12), was diagnosed simultaneously (n = 11), but never preceded LCH. The BRAF-V600E mutation was found in 11/16 LCH lesions (69%) and 9/11 ECD lesions (82%). These findings indicate that the association of LCH and ECD is not fortuitous, and suggest a link between these diseases involving the BRAF-V600E mutation.
Berres et al recently proposed that LCH could be redefined as an inflammatory myeloid neoplasia.39 There are, indeed, genetic, molecular and functional data implicating ERK signaling pathway activation at critical stages of myeloid differentiation, as an essential and universal driver of LCH. Given the high frequency of associations between LCH and ECD and the similar mutations affecting the MAP kinase pathway found in both conditions, we also speculate that ECD could be redefined as an inflammatory myeloid neoplasia.
Diagnostic criteria
The diagnosis of ECD is based on histopathologic findings with the appropriate clinical and radiological context. Lesional tissue usually shows foamy or lipid-laden histiocytes with surrounding fibrosis or xanthogranulomatosis; Touton giant cells are often present. Immunohistochemistry shows histiocytes that are factor XIIIa+/CD68+/CD163+/fascin+ and CD1a−/Langerin−/Birbeck granules−. Positivity for S100 has been observed rarely. The radiographic finding of bilateral and symmetric diaphyseal and metaphyseal osteosclerosis in the legs is present in almost all patients. This is best seen on bone scan (99Technetium) which reveals radiotracer uptake by the distal ends of the femurs and the proximal and distal tibia (in 96% of a series of 53 patients from 2011),40 and less sensitively by positron emission tomography (PET). Bone lesions may be missed on plain films but can be visualized on computed tomography (CT) or magnetic resonance imaging (MRI). Abdominal CT may show dense infiltration of perinephric fat, or “hairy kidney”, in 57% of cases. A biopsy is always necessary to establish the diagnosis of ECD, and when “hairy kidney” aspect is present, a CT-guided biopsy of the perirenal infiltration is the recommended approach.41 Biopsy of skin lesions, such as xanthelasmas, are also feasible. Biopsy is also helpful to establish the BRAF mutational status, which has major therapeutic implications.
Clinical features
ECD is a true multisystemic disease, as virtually all organs can be involved. Patients may present with skeletal, pulmonary, retroperitoneal, endocrine, neurologic, skin, renal, and cardiovascular involvement.31,33 The extent and distribution of the disease determine the clinical course. Some cases present with only asymptomatic bone lesions, whereas others have multisystemic, potentially life-threatening disease.
Skeletal involvement of the long bones is almost universal (96% of 53 patients included in the 2011 series), but only 39% of patients suffer bone pain, which is the most common clinical feature of ECD.42 Bone pain is usually mild, may start at any time during the course of the disease and mostly affects the distal limbs. This is in contrast to LCH, which typically involves the calvarium, facial bones, proximal limbs, pelvis, and scapula. Cardiovascular involvement is very common (70%), but may be asymptomatic and detected incidentally by heart MRI or CT. The most frequent cardiovascular abnormality is circumferential soft tissue sheathing of the thoracic or abdominal aorta (55% and 57% of cases, respectively).31,33 The whole aorta can be sheathed on CT scan, the so called “coated aorta”, and this has become one of the iconic features of ECD (43% of cases).42 Periarterial infiltration may extend to the main aortic branches. Its clinical consequences are usually not severe, except for renovascular hypertension (16% of cases) which may require stenting.33
Pericardial disease is frequent (30%) and can present as pericarditis, effusion or even tamponade.33 Pseudotumoral infiltration of the right atrium is present in nearly a third of the patients, and is best visualized on MRI.43
Orbital infiltration, often bilateral, occurs in 25% of patients and can present with exophthalmos, retro-oribital pain, oculomotor nerve palsies, or blindness. This infiltration may be significant and refractory in a small number of cases, and may require surgical debulking. Xanthelasma of the eyelids or periorbital tissues occurs in 25% of ECD patients.
Diabetes insipidus, due to pituitary gland infiltration, can develop in 26% of ECD patients but hyperprolactinemia, gonadotropin insufficiency, and hypotestosteronism may also occur. Approximately one-third of ECD cases present with pseudo “retroperitoneal fibrosis”, in some cases complicated by bilateral hydronephrosis, which may require ureteral stenting.44 Unlike idiopathic retroperitoneal fibrosis, pelvic ureters, and inferior vena cava are usually not affected in ECD.33
The frequency of CNS involvement in ECD patients varies between 25% and 40%.42,45 Cerebellar and pyramidal syndromes are the most frequent neurological signs, although some patients may present with seizures, headaches, neuropsychiatric symptoms, cognitive impairment, sensory disturbances, cranial nerve paralysis, or asymptomatic lesions. Neurological involvement may eventually lead to severe functional disability particularly when facing cerebellar involvement. CNS involvement is a major prognostic factor in ECD, as survival analysis has identified this factor as an independent predictor of death (hazard ratio = 2.51; 95% confidence interval, 1.28-5.52; P = 0.006).31 The most devastating and resistant neurological complication is neurodegenerative cerebellar disease, which is present in 17% of ECD patients.
The clinical course of ECD is that of a chronic disease, although it has not been described in detail yet. ECD Lesions tend to accumulate in the affected organs and rarely regress spontaneously. Serum C-reactive protein (CRP) levels are high in >80% of cases,42 but with little impact on treatment outcome. Disease activity in ECD patients is assessed by regular clinical examinations and radiological investigations (every 6 months) to assess for morphological changes or response to therapy. FDG-PET scans are particularly informative for the assessment of ECD activity. Follow-up PET scans can detect CNS involvement, and can detect rapid early response to therapy better than MRI. PET scans can also be used to investigate the cardiovascular system. The pilot study on the use of BRAF inhibitors in ECD patients illustrates the value of PET scan in monitoring for disease activity.46
Treatment
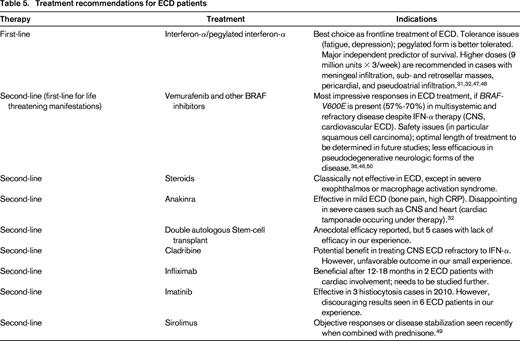
Before 2005, or prior to the first report showing benefit from IFN-α as ECD treatment, many therapeutic regimens were reported including steroids, cytotoxic agents, vinca-alkaloids and double-autologous hematopoietic stem cell transplantation. The efficacy of these regimens is difficult to establish, due to the small number of patients, heterogeneous treatment, and short follow-up time. Braiteh et al reported rapid, marked, and persistent regression of retro-orbital infiltration and a progressive improvement of bone lesions, pain, and diabetes insipidus in 3 ECD patients given IFN-α.47 However, with a larger study showed that efficacy of low-dose IFNα (3 MU X 3/week) differed between the sites involved.48 In some cases, the symptoms failed to respond to such low doses of IFNα; this was particularly true in patients with severe multisystem forms of ECD (CNS and cardiovascular involvement).31 We therefore recommend higher doses, 9 MU X 3/week if possible, because such doses may be more effective against meningeal infiltrations, sub- and retrosellar masses, and pericardial and pseudoatrial infiltrations. However, IFN-α may lead to adverse effects, including depression, fatigue, myalgias, fever, gastrointestinal symptoms, and myelosuppression. IFN-α treatment has also given disappointing results in cases of neurodegenerative cerebellar involvement (similar to that observed in LCH). Nevertheless, IFN-α appears to be the best choice for the initial prolonged treatment of ECD. Survival analysis from a series of 53 patients indicated that treatment with IFNα and/or PEGylated IFNα was a major independent predictor of survival (HR = 0.32; 95% CI, 0.14-0.70; P = 0.006).31 We generally begin treatment with PEGylated forms of IFN-α because such forms are better tolerated than IFN-α. The dosage is usually determined by severity and organ dominance of disease.
Cladribine, a nucleoside analog, has been used in the treatment of newly diagnosed and refractory ECD, but anecdotal reports of its efficacy are few.32 The treatment of 2 ECD patients (neither with cardiovascular or CNS involvement) with recombinant human IL-1 receptor (anakinra) appeared to be promising. We therefore treated 12 ECD patients at our institution with anakinra. Treatment efficacy was poor overall, particularly for the severe forms of the disease (cardiovascular and CNS involvement). Other groups have shown this treatment to be effective only for mild ECD (mostly bone pain). Infliximab treatment was shown to be beneficial after 12-18 months, in 2 ECD patients with cardiac involvement. Recently, sirolimus combined with prednisone was reported in an open-labeled trial with 10 patients showing disease stabilization or objective responses.49
Because some lesions in ECD have abundant expression of PDGFR-β, treatment with imatinib was thought to be a reasonable strategy as second-line therapy. Results in 7 ECD patients treated with imatinib are nevertheless variable, and seem to be more effective in less-advanced disease. Radiotherapy to ECD has been reported but mainly as short-term palliation and should therefore be avoided. Surgical debulking is limited in ECD to severe orbital lesions or resectable intracranial lesions.32
However, it was the demonstration in 2012 of efficacy for a BRAF inhibitor (vemurafenib) that was the most promising advance. That year, we conducted a pilot study of vemurafenib treatment for 3 patients with multisystemic and refractory ECD who carried the BRAF-V600E mutation. Two of the patients also had skin or lymph node LCH.46 Vemurafenib treatment led to rapid, substantial clinical and biological improvement in all 3 cases. We subsequently treated 5 other patients with this drug, for which we recently reported long-term efficacy.36,50 One patient developed squamous cell carcinoma after 6 months of treatment, but no other major adverse effects were reported in the others. We therefore recommend considering vemurafenib treatment for all patients with severe and refractory BRAF-V600E mutated ECD, particularly if life threatening. The importance of BRAF inhibition has been confirmed and extended to larger series of patients, both by our group and by many other groups worldwide, including that of the MSKCC. BRAF inhibitors have been used to treat >40 patients in total since 2012 (J.H., personal communication at the second ECD Medical Symposium in Bethesda, September 2014).
In 2013, Diamond et al described 1 patient with ECD and NRAS mutation.51 We think it's likely that ECD patients with NRAS mutations would also benefit from targeted anti-MEK therapy, as have some patients with metastatic melanomas. Moreover, the possible benefits of combined treatment (anti-MEK & anti-BRAF) should also be investigated in histiocytoses, as this approach might be more effective and less toxic, as already demonstrated in melanoma patients.
The survival analysis of 122 patients followed in our center in 2014 indicated that overall mortality following treatment with IFNα and, more recently, vemurafenib, was only 22%, with a 5 year survival of 82.8% (J.H., personal communication at the second ECD Medical Symposium in Bethesda, September 2014).
Conclusions
The recent discoveries of recurrent mutations of the MAP kinase and PI3K pathways in JXG and ECD, the observation that BRAF-V600E mutations can be identified in myeloid progenitor cells and the observed clinical responses to BRAF/MAPK inhibition support the redefinition of ECD and JXG as inflammatory myeloid neoplasms, similarly to LCH. The increase in the diagnosis rate of ECD is due mostly to a greater awareness among pathologists, radiologists, and clinicians of the various aspects of this previously obscure disease. Substantial progress has been made in recent years, such as the beneficial effect of IFN-α treatment, and the demonstration that BRAF inhibition is highly effective in severe cases of BRAF-V600E-mutated ECD. Further studies on this disease and a better understanding of its pathogenesis should lead to the development of more effective targeted therapies. New and more tolerable BRAF inhibitors, as well as a combination of BRAF and MEK inhibitors should be investigated in the future for patients with severe BRAF-mutated ECD and refractory disease.
Future larger scale WES studies of JXG lesions may lead to the discovery of activating kinase mutations involving BRAF-V600E, ARAF, or MAP2K1, which could open the door to new effective and tolerable targeted therapies for patients with severe systemic JXG. Due to the rarity of RDD and other non-LCH disorders, prospective treatment studies can be a challenge to conduct. Therefore, the Histiocyte Society has recently opened an international registry for the rare histiocytic disorders or IRHDR (ClinicalTrials.gov; NCT02285582) to better understand the natural history, clinicopathologic features and the most effective therapeutic strategies for the non-LCH disorders.
Correspondence
Julien Haroche, UPMC Univ Paris 06, Institut E3M, Pitié-Salpêtrière Hospital, 47–83 Boulvevard de l'Hôpital, 75651 Paris, France; Phone: + 33 1 42 17 80 37; Fax: + 33 1 42 16 58 04; e-mail: julien.haroche@psl.aphp.fr.
