
Abstract
Randomized clinical trials (RCTs) have determined, in surgical and critically ill patients, relatively safe hemoglobin (Hb) thresholds of 7-8 g/dL to guide restrictive transfusion of red blood cells (RBCs). However, in patients with various hematologic disorders, strong evidence in support of such an approach is sparse and the optimal transfusion practice is yet to be defined. This review focuses on RBC transfusion practice in three hematologic diseases and a treatment strategy, including autoimmune hemolytic anemia, thalassemia, myelodysplastic syndrome, and hematopoietic stem cell transplantation. These entities manifest in a broad spectrum of anemia, acute or chronic, in patients with different comorbidities and degrees of transfusion requirement. Thus the nuances in the indications of RBC transfusion and the goals to achieve in these specific situations may have been underappreciated. The limited data available highlight the importance of titrating RBC transfusion based on the clinical context and patient characteristics. Future RCTs are necessary to firmly establish the Hb thresholds associated with improved outcomes relevant to these specific patient populations, which will facilitate the personalized decision-making in RBC transfusion.
Learning Objectives
To understand the evidence supporting the current transfusion practice and its limitations in patients with hematologic disorders and anemia resulting from therapy
To understand the goal(s) of red blood cell transfusion in patients with representative hematologic disorders and anemia resulting from therapy
Transfusion of red blood cells (RBCs) is a critical intervention in patients with symptomatic anemia. Although multiple mechanisms allow tissue oxygenation to be maintained within a broad range of hemoglobin (Hb) levels, a high mortality rate has been observed when the Hb is <6 g/dL in the setting of acute blood loss.1 On the other hand, RBC transfusion is generally believed, in both adults and children, to be unnecessary when the Hb is >10 g/dL. It is within this gray zone between Hb levels of 6 and 10 g/dL that questions frequently arise whether to transfuse a patient or not. With the increasing awareness of the hazards inherent to blood transfusion, potential benefits from a transfusion should always be weighed against the potential risks. Considering the sheer number of RBCs transfused, >13 million units annually in the United States, even rare reactions may harm a sizable number of patients.
The 2012 AABB Guideline on RBC transfusion recommended a restrictive transfusion strategy in hospitalized stable patients based on a systematic review of available randomized clinical trials (RCTs).2 The patients transfused liberally did not have improved mortality or other outcomes examined, despite having an average pretransfusion Hb level of 1.48 g/dL higher than those treated with a restrictive transfusion strategy. In contrast, the restrictive strategy significantly decreased the number of transfused patients (328 fewer per 1000 patients) and blood utilization. It was recommended that, in hospitalized hemodynamically stable patients, RBC transfusion should be considered at a Hb level of 7-8 g/L or less, or for symptoms including orthostatic hypotension or tachycardia unresponsive to fluid resuscitation, chest pain, or congestive heart failure. For patients with preexisting cardiovascular diseases, a threshold of 8 g/L or less was recommended. No recommendation was made for patients with acute coronary syndrome due to lack of data. The advantage of restrictive transfusion continued to be solidified with emerging evidence from patients with septic shock3 and head injury,4 among others,5 although a trial in patients undergoing cardiac surgery created a new uncertainty by showing a higher all-cause mortality with the restrictive transfusion group.6
Despite the positive impact of the AABB guidelines on RBC transfusion practices, there have been some legitimate criticism of the clinical trials from which the guidelines were derived. First, available RCTs did not compare the restrictive transfusion strategy to the current standard of care of titrating transfusions based on the conditions of individual patients. Moreover, when fixed transfusion thresholds were examined in an RCT, subgroups of patients may be overtransfused or undertransfused against the standard of care, causing “practice misalignment” and potentially confounding the conclusion.7 Finally, blood products in an era of universal leukoreduction and elaborate donor testing may have different risk profiles than a decade ago when some of the key evidence was generated. Notwithstanding these caveats, Hb concentration remains the most important clinical parameter on which physicians base their decision to transfuse patients, and there are technical challenges to incorporate titrated transfusion practice into a control arm. In the long run, some of the controversies may be resolved with additional trials, and future guidelines will continue to absorb the best evidence available.
The practice of blood transfusion perhaps needs to be personalized rather than standardized, especially for a myriad of hematologic disorders underlying a broad spectrum of anemia. Previous RCTs primarily focused on surgical or hospitalized critically ill patients with relatively acute anemia (Figure 1). Although “off-label” use of a restrictive transfusion strategy may be common in certain patient populations in the absence of high-quality evidence, it is important to recognize that the goal for RBC transfusion varies in many hematologic disorders, and challenges unique to these disorders may affect the efficacy and safety of blood transfusion. In this review, we are going to focus on 3 hematologic diseases and a treatment strategy, including autoimmune hemolytic anemia (AIHA), thalassemia, myelodysplastic syndrome (MDS), and hematopoietic stem cell transplantation (HSCT), to illustrate and contrast the diverse clinical contexts in which RBC transfusion is indicated. Due to the scarcity of RCTs in these areas, our conclusions will be based on available trials, observational studies, and expert opinions.

The evidence-base of RBC transfusion strategies. Patients enrolled in 32 RCTs are grouped into nine major categories (in italic bold font). Detailed patient characteristics in individual trials are omitted for simplicity. Each RCT is labeled with trial acronym or the first author and year of publication. Limited space does not allow listing all references, but all studies except TITRe26 and PINT38 have been included in a recent meta-analysis.5 *Pediatric patients; **extremely low birth weight infants; †exclusion criteria of the study explicitly mentioned certain hematologic conditions (eg, any preoperative anemia, chronic anima, or hemolytic anemia).
The evidence-base of RBC transfusion strategies. Patients enrolled in 32 RCTs are grouped into nine major categories (in italic bold font). Detailed patient characteristics in individual trials are omitted for simplicity. Each RCT is labeled with trial acronym or the first author and year of publication. Limited space does not allow listing all references, but all studies except TITRe26 and PINT38 have been included in a recent meta-analysis.5 *Pediatric patients; **extremely low birth weight infants; †exclusion criteria of the study explicitly mentioned certain hematologic conditions (eg, any preoperative anemia, chronic anima, or hemolytic anemia).
RBC transfusion for selected hematologic disorders
Autoimmune hemolytic anemia
AIHA is caused by autoantibodies that destroy the patients' own RBCs. In vitro, these antibodies appear to be panagglutinins because they agglutinate all reagent RBCS and autologous cells when tested against a panel of RBCs. Depending on the direct antiglobulin test (DAT) results, the thermal range of the autoantibody and presumed immunoglobulin class driving the disease, AIHA can be classified into warm, cold, or mixed types. Warm AIHA is routinely treated with steroids. Rituximab and splenectomy have been used as second line options. Cold AIHA requires avoidance of cold exposure and has been shown to respond well to rituximab. Mixed AIHA frequently present with lower Hb levels and may benefit from aggressive treatment, such as rituximab.8 If AIHA is secondary to drugs, malignancy or other immune diseases, the underlying condition must be treated in addition to the effort to halt hemolysis. Critical to all AIHA types is the initial and ongoing evaluations of the need for RBC transfusion.
The goal of RBC transfusion in AIHA patients is to sustain basic tissue oxygenation before the medical treatments take effect. This apparently simplistic goal is complicated by several challenges unique to AIHA. First, all RBC units will appear “incompatible” at crossmatch due to the presence of a panagglutinin. When informed about such findings, physicians may be “intimidated” and withhold even necessary transfusions. Careful evaluation of previous transfusion and pregnancy history is essential in determining the risk of transfusions in this situation. Second, higher rates of alloimmunization to RBC antigens have been observed in patients with AIHA. Once transfused, close monitoring for alloimmunization is required, necessitating complicated serologic workup prior to future transfusions. Because autoantibodies interfere with the detection of underlying alloantibodies, the serologic testing for AIHA patients incurs the highest per patient cost compared to patients with other diagnoses.9 In addition these complicated workups increase the turnaround time to transfusion. There may well be increased risk of hemolysis due to missed alloantibodies. Third, transfused RBCs bring only temporary benefits, as autoantibodies shorten the survival of both autologous and allogeneic blood. If a significant portion of hemolysis is intravascular, excessive transfusion may endanger kidney function and aggravate systemic inflammation. Finally, patients may present with a spectrum of disease severity and functional reserve, and there is no objective biomarker to predict whether or how soon a patient will respond to treatment. Therefore, it is difficult to gauge the benefit of immediate RBC transfusion versus watchful waiting.
The presenting Hb levels in a large cohort of patients with AIHA were reported recently.8 The majority of the patients with warm and cold AIHA presented with Hb levels >6 g/dL, whereas Hb concentrations were <6 g/dL in most patients with mixed AIHA or atypical AIHA (Figure 2). Only 115 (37%) patients were actually transfused in this cohort, and 38 (33%) of those transfused did not achieve the expected efficacy defined as a Hb increment of 1 g/dL per unit transfused and remaining stable for ≥3 days. Importantly, more patients with severe onset AIHA (43% with Hb ≤6 g/dL) did not respond to transfusion, suggesting that those who need transfusion most are less likely to benefit from it.

Presenting hemoglobin levels (g/dL) in 308 patients with primary AIHA. The grouped histogram is a visualization of data presented by Barcellini et al.8 Different AIHA types were defined as follows: warm AIHA: DAT+ for IgG (with or without C3d); cold AIHA: DAT+ for C3d only with I-specific cold agglutinins at titer ≥64; mixed AIHA: DAT+ for IgG and C3d, with coexisting warm and high-titer cold autoantibodies; atypical AIHA: DAT−, or DAT+ for IgA only, or warm IgM, or mitogen-stimulated DAT+ only.
Presenting hemoglobin levels (g/dL) in 308 patients with primary AIHA. The grouped histogram is a visualization of data presented by Barcellini et al.8 Different AIHA types were defined as follows: warm AIHA: DAT+ for IgG (with or without C3d); cold AIHA: DAT+ for C3d only with I-specific cold agglutinins at titer ≥64; mixed AIHA: DAT+ for IgG and C3d, with coexisting warm and high-titer cold autoantibodies; atypical AIHA: DAT−, or DAT+ for IgA only, or warm IgM, or mitogen-stimulated DAT+ only.
Due to the lack of evidence to guide transfusion for AIHA, no specific Hb threshold can be recommended. Although it is tempting to equate the AIHA patient population with the intensive care unit (ICU) patients enrolled in the RCTs listed in Figure 1, the risk and benefit profile of RBC transfusions in AIHA patients has shifted, being inherently riskier and transiently beneficial, as mentioned above. Nevertheless, patients with AIHA are unlikely to benefit from a liberal transfusion strategy and may well be harmed if ongoing hemolysis is fueled by excessive transfusions.10 Perhaps the ultimate question for AIHA is how low the Hb level can be allowed to slip before a transfusion is really needed. The answer will eventually be based on the symptoms and risk factors of a patient regardless of the Hb levels, and physicians should not feel obligated to transfuse when Hb is lower than a value specified by any guidelines. Blood transfusion may only be needed in situations when a patient has neurological symptoms, or experiences chest pain, or has rapidly progressing anemia, or develops early signs of heart failure.11 The dose of transfusion should be one unit at a time in adults and 10 mL/kg in children followed by assessing whether the symptoms have improved.
Thalassemia
Thalassemia is an inherited form of anemia in which genetic abnormalities reduce or abolish the production of α- or β-globin chains. The imbalanced synthesis of α- and β-globin chains leads to ineffective erythropoiesis and chronic hemolytic anemia. The severity of thalassemia syndromes is dictated by the particular combination and extent of genetic aberrations, presence of other structural Hb variants (eg, HbS and HbE), and influences from additional genetic and environmental modifiers. We include thalassemia in this review because RBC transfusion is essential to the care for thalassemia patients. Moreover, this patient population is predominantly pediatric and ambulatory. It is of paramount importance to revisit the goals of RBC transfusion and the standard of care in this setting. The transfusion practices in another closely related entity, sickle cell disease, has been extensively covered in recent ASH education programs, and is therefore omitted.
Thalassemia syndromes can be phenotypically classified into transfusion dependent and nontransfusion dependent thalassemias (TDT & NTDT). The former requires lifelong regular transfusions and is frequently associated with β-thalassemia major (β-TM), severe HbH disease or severe HbE/β-thalassemia.12 NTDT syndromes encompass β-thalassemia intermedia, mild HbH disease, or mild to moderate HbE/β-thalassemia among others, and affected patients do not rely on transfusions for survival.13 However, there is no absolute correlation between the phenotypes and genetic abnormalities (Figure 3).14 NTDT could be misclassified as TDT if regular transfusions were initiated prematurely and no attempt was made to withdraw or taper the regimen.

Diagnosis and transfusion regimens in 407 thalassemia patients enrolled in the Centers for Disease Control and Prevention (CDC) Thalassemia Blood Safety Network. The plot is a visualization of data presented by Vichinsky et al.14 The width of the ring is proportional to the total number of patients in each category. Thalassemia major (TM) patients in the “never transfused” category were under 1 year of age and yet to begin chronic transfusion at the time of enrollment. TI indicates thalassemia intermedia; and Thal, thalassemia.
Diagnosis and transfusion regimens in 407 thalassemia patients enrolled in the Centers for Disease Control and Prevention (CDC) Thalassemia Blood Safety Network. The plot is a visualization of data presented by Vichinsky et al.14 The width of the ring is proportional to the total number of patients in each category. Thalassemia major (TM) patients in the “never transfused” category were under 1 year of age and yet to begin chronic transfusion at the time of enrollment. TI indicates thalassemia intermedia; and Thal, thalassemia.
The goals of RBC transfusion in thalassemia are multifaceted. Not only is transfusion crucial for survival in TDT, it also ensures proper growth and development in pediatric patients. Transfused RBCs further suppress ineffective erythropoiesis, attenuate extramedullary hematopoiesis, and prevent complications such as splenomegaly, skeletal abnormalities, gallstones, increased absorption of dietary iron, and thrombosis. This usually requires more intensive transfusions and almost inevitably leads to iron overload in the absence of chelation therapy. Thus, the transfusion goals in thalassemia patients are distinct from those in adult patients with relatively acute anemia, for whom alleviation of anemia can be achieved with a more restrictive transfusion strategy in most cases.
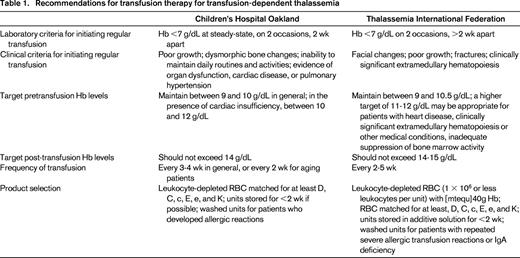
The targets of transfusion have been outlined in several practice guidelines, and the recommendations from the United States (Children's Hospital, Oakland, CA)15 and the Thalassemia International Federation (TIF)12 are summarized in Table 2. Both guidelines emphasize the importance of clinical assessment over anemia alone (Hb <7 g/dL), and the recommended target pretransfusion Hb levels are almost identical (9-10 vs 9-10.5 g/dL). Upper limits of post-transfusion Hb levels are also specified to prevent over transfusion. The TIF guidelines specify broader indications for a higher pretransfusion Hb target between 11-12 g/dL. In addition to heart disease, the higher target is recommended for clinically significant extramedullary hematopoiesis or other medical conditions, inadequately suppressed bone marrow activity, as well as symptoms of back pain prior to blood transfusion. These recommendations, however, may increase the number of patients on escalated transfusion regimens. For NTDT, TIF constructed a separate set of guidelines based on observational studies and expert opinions, which specified a much lower transfusion threshold (Hb <5 g/dL) and emphasized the importance of tailoring the transfusion frequency to suite different clinical morbidities and contexts.13
Two studies are frequently referenced to support the recommendation on pretransfusion Hb targets for thalassemia. First, in patients with β-thalassemia major, pretransfusion Hb levels were inversely related to serum erythropoietin and transferrin receptor.16 Using transferrin receptor as an indicator for erythropoiesis, a pretransfusion Hb of 9-10 g/dL correlated with an erythroid activity 1-4 times normal, which was comparable to most subjects with β-thalassemia trait. Second, in a study comparing 32 β-TM patients receiving moderate transfusion (pretransfusion Hb within 9-10 g/dL) to the same group of patients receiving hypertransfusion (pretransfusion Hb within 10-12 g/dL) in the past as historical controls, moderate transfusion successfully maintained the erythroid activity within 0.8-4 times normal while reducing mean blood consumption by 33 mL RBC/kg/year and mean serum ferritin by 52%.17 Despite the above evidence, not a single RCT has been performed to validate or improve the practice of blood transfusion in this population.
Recent reports offered a peek into the actual transfusion practice in the United States. Among 407 thalassemia patients, with a median age of 31 years (range, <1-58 years), followed from 2004 through 2012 in the Center for Disease Control and Prevention (CDC) Thalassemia Blood Safety Network,14 80% (327) had been transfused chronically. Nine percent (38) had been transfused intermittently, and 10% (42) never transfused. The proportions of different diagnoses in the 3 categories are illustrated in Figure 3. In terms of complications, iron overload and end organ damage requiring medical treatment were common despite 78% of patients receiving chelation therapy. Splenectomy was reported in 45% of patients,14 implying a high prevalence of hypersplenism possibly secondary to inadequate transfusion.
A separate survey of 8 thalassemia treatment centers18 reported that most centers transfuse patients when the Hb level is <8, 9, or 10 g/dL, whereas 1 center reported that transfusion is purely patient dependent. The amount of RBCs transfused per kilogram body weight (pediatric patients) or per episode is also highly variable. The criteria to initiate or withdraw chronic transfusions, target pretransfusion Hb, and frequency of transfusions were not captured in this survey. Selection of blood products was also variable.18 Only 3 centers provided units within 14 or 21 days of collection in compliance to current guidelines (Table 1). Only one-half of all centers preventively matched for Rh (C, c, E, e) and K antigens, which is not in line with current guidelines12,15 and may partly account for the alloimmunization rate of 19% among transfused patients in the CDC cohort.14
Myelodysplastic syndrome
MDS is a heterogeneous group of diseases characterized by dysplasia in one or more myeloid lineages, associated cytopenia, and a risk of progression to acute leukemia. Most patients with MDS will develop symptomatic anemia and require RBC transfusions at some point in their clinical course. Depending on the risk categories defined by the International Prognostic Scoring System (IPSS), from low to high risk, 39%-79% of patients were reported to be RBC transfusion-dependent.19 Transfusion dependency is also a harbinger of poor prognosis, which could be explained by more advanced disease, worse anemia, and transfusion-related complications. On the other hand, decreased transfusion requirements are an indicator of good response to other therapeutic agents (see below).
Some clinical factors may impact the planning of RBC transfusions for MDS patients. First, patients are diagnosed with MDS at a median age of 70 years,20 which is at the opposite end of the spectrum from patients with thalassemia. This population has a high prevalence of cardiac disease and other comorbidities, and may not tolerate anemia as well as younger patients. Second, transfusion requirements can be profoundly affected by other treatments as well as disease progression. Some patients may respond to erythropoietin, immunomodulatory agents or chemotherapy,21 and transfusions should be adjusted accordingly based on clinical assessment. Conversely, when patients lose the response to a treatment, transfusions must be scaled up as needed. Third, blood transfusions are ultimately a palliative measure for MDS, which should aim at relieving anemia-related symptoms and improving the quality-of-life (QOL).
Current guidelines for MDS by the National Comprehensive Cancer Network (NCCN)22 recommend RBC transfusions for symptomatic anemia only. Transfusion should be avoided for arbitrary Hb thresholds in the absence of symptoms of active coronary disease, heart failure, or stroke. Only the smallest effective dose is recommended to relieve symptoms of anemia or to return the patient to a safe Hb level (7-8 g/dL in stable, noncardiac in-patients). These recommendations did not emphasize anemia-related symptoms such as excessive fatigue, which could severely impact the QOL and has been reported in >90% of patients with MDS.23 A correlation between fatigue scores and Hb levels has also been demonstrated in transfusion-dependent patients. Because most patients with MDS are ambulatory, transfusions at regular intervals may help establish a routine, and ideally the dose of transfusion should be titrated to maintain a Hb level that correlates with acceptable QOL for each individual. Of note, to improve QOL was included in the guidelines from the United Kingdom24 and the European LeukemiaNet (ELN)25 as an objective of RBC transfusion for MDS patients. ELN also made a general recommendation to transfuse patients with severe anemia (Hb <8 g/dL) or milder anemia with symptoms, although the optimal Hb level is unknown.
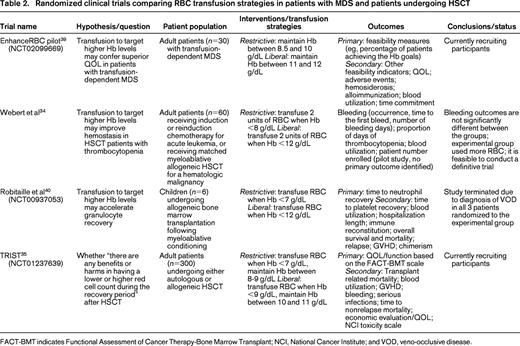
It remains unclear to what extent RBC transfusions can improve QOL in MDS patients and under what conditions. Studies of blood transfusion and QOL in MDS patients have been hampered by small samples, diverse transfusion protocols, and poor timing of QOL assessment.26 There is also a lack of consensus instrument for QOL measurement, although some tools are gaining favor, such as the European Organization for Research and Treatment of Cancer Core Quality-of-Life (EORTC QLQ-C30). Eventually the impact of different transfusion strategies on QOL needs to be investigated in prospective randomized trials using validated questionnaires. One such trial (EnhanceRBC; Table 2)27 will randomize transfusion-dependent patients to a liberal transfusion arm where Hb will be maintained between 11 and 12 g/dL, and a restrictive transfusion arm where Hb will be maintained between 8.5 and 10 g/dL. If the ongoing pilot study proves feasible, the following expanded trial will be able to definitively compare changes in QOL scores between the 2 arms. Despite a long way to go, this type of study is necessary to better define the optimal RBC transfusion practice in MDS patients.
Randomized clinical trials comparing RBC transfusion strategies in patients with MDS and patients undergoing HSCT
FACT-BMT indicates Functional Assessment of Cancer Therapy-Bone Marrow Transplant; NCI, National Cancer Institute; and VOD, veno-occlusive disease.
Finally, patients with MDS share the same long-term sequela of chronic RBC transfusion with thalassemia patients. Iron chelation may reduce iron overload and has been associated with better survival in a recent meta-analysis of 8 observational studies.28 However, the definitive benefits of chelation therapy still await confirmation by prospective randomized trials. High alloimmunization rates (15%) have also been reported in chronically transfused patients with MDS, and RBCs phenotypically matched for Rh (C, c, E, e) and K could be beneficial in this population although the cost-effectiveness has not been proven.29
Hematopoietic stem cell transplantation
Transfusion of RBCs, platelets and sometimes granulocytes provides essential support for patients undergoing HSCT before sufficient hematopoiesis is achieved in all lineages. The current approach to platelet transfusions is supported by multiple randomized trials comparing the relative efficacy and safety of therapeutic versus prophylactic platelet transfusions, as well as different dosing strategies.30 In contrast, there is a paucity of data to define the optimal Hb levels to target following HSCT. A recent survey of 15 adult transplant centers in Canada found that 14 centers routinely transfuse 2 units of RBC when the Hb level is <8 g/dL, whereas 1 center uses a threshold of 7 g/dL.31 A similar trend was found among pediatric transplant centers with 60% using a Hb level of 8 g/dL and 25% using a Hb level of 7 g/dL.32 The rationale for the majority of centers to adopt a threshold of 8 g/dL remains unclear, yet the practice mimics the restrictive transfusion practice in ICU patients.
It must be recognized that there are differences between HSCT and ICU patients, although both are critically ill and heavily transfused. HSCT patients may have additional risk factors, such as advanced hematologic malignancy, severe anemia prior to HSCT, aggressive conditioning regimens, and major ABO incompatibility with allogeneic donors. Many of these factors have been associated with increased and prolonged transfusion requirement in retrospective studies.33 In addition, HSCT patients have cytopenias in multiple lineages rather than simple anemia, and they are prone to severe infections due to profound immune deficiency. Despite these differences, it is unclear whether HSCT patients are likely to do better with a more liberal transfusion strategy or a restrictive transfusion strategy.
Several past and ongoing RCTs were designed to examine three different aspects of the same question: is it beneficial to target a higher Hb level in HSCT patients (Table 2)? Webert et al focused on the interaction between anemia and thrombocytopenia following chemotherapy or myeloablative HSCT and the associated bleeding risk.34 Sixty adult patients were randomized in this pilot study. The liberal transfusion strategy (Hb threshold = 12 g/dL) used more RBCs per patient but did not improve bleeding outcomes compared with the restrictive strategy (Hb threshold = 8 g/dL). Despite a limited power to detect small differences, this pilot study did prove feasible and could be developed into a definitive trial. Robitaille et al set out to examine whether targeting a higher Hb level may accelerate granulocyte recovery in pediatric HSCT patients. Unfortunately, the study was terminated after randomization of 6 patients due to severe vaso-occlusive disease (VOD) in all 3 patients in the liberal transfusion arm (Hb threshold = 12 g/dL). None of the 3 patients in the restrictive arm (Hb threshold = 7 g/dL) was affected. This complication was not reported in adult patients subjected to the same liberal transfusion strategy.34 It is possible that liberal transfusions led to exacerbated inflammation, increased viscosity, and a procoagulant state, but it remains unclear why these effects manifested in pediatric patients only. Finally, an ongoing trial (TRIST; transfusion of red cells in HSCT) will compare the potential benefits and harms associated with liberal (Hb threshold = 9 g/dL) versus restrictive (Hb threshold = 7 g/dL) transfusion strategies in 300 HSCT patients.35 The primary outcome is the QOL or functionality as measured by FACT-BMT (Functional Assessment of Cancer Therapy-Bone Marrow Transplant). Secondary outcomes including transplant related mortality and blood utilization will also be compared.
It is important to note that the role of transfusion support in HSCT patients will continue to evolve given the excellent results from RCTs evaluating the efficacy of erythropoietin therapy following both allogeneic36 and autologous37 HSCT. Once the optimal regimen of erythropoietin and its safety are firmly established, RBC transfusion will likely be significantly reduced in this population and the period of transfusion support shortened. If this occurs in the near future, a restrictive transfusion practice may suffice.
Conclusion
Current evidence supports a restrictive transfusion practice in most surgical and critically ill patients with relatively acute anemia. However, patients with various hematologic disorders have not been the focus of RCTs and the approach to RBC transfusion in these patients remains largely empirical. A brief review of the transfusion practices in 3 hematologic diseases and a treatment strategy revealed a landscape of immense complexity due to the diverse natures of anemia, different levels of transfusion dependency and shifting risk-benefit profiles of transfusion therapy in these various situations. Patients with AIHA should only be transfused in presence of symptoms and signs consistent with decompensating anemia. Because RBC transfusion is challenging with a higher risk-benefit ratio in these cases, careful clinical evaluation should override the commonly used Hb threshold of 7 g/dL in deciding whether to initiate RBC transfusion. In contrast, for patients with severe thalassemia, the question is frequently whether these younger patients are adequately transfused in order to suppress ineffective erythropoiesis and to ensure normal growth and development. In the absence of any new high-quality evidence, the current guidelines on the transfusion practice for thalassemia still stand (Table 1). Prospective trials are necessary to validate the current recommendations or to evaluate possible alternatives. For patients with MDS, all established and novel therapeutic options should be explored to reduce or delay transfusion dependency. The need for occasional or regular transfusions should be determined based on the overall clinical picture, including a patient's age, comorbidities, Hb levels, and severity of anemia-related symptoms, whereas no specific Hb target range can be recommended at this point. Regarding the long-term complications of transfusion in both thalassemia and MDS, extended antigen matching can effectively mitigate the high rate of alloimmunization in these patients but are not universally done perhaps due to lack of resources; the quantitative interactions among RBC transfusion, iron overload and iron chelation therapy also need to be clarified. Finally, in patients undergoing HSCT, a transfusion threshold at a Hb level of 7 or 8 g/dL is well accepted among transplant centers, which mirrors the current AABB guidelines for critically ill, hospitalized patients. There is no evidence to support targeting higher Hb levels in this population, but ongoing clinical trials are examining its impact on various outcomes including QOL.
Correspondence
Brenda J. Grossman, Department of Pathology and Immunology, Washington University School of Medicine, 660 S Euclid Ave, Box 8118, St Louis, MO 63110; Phone: 314-362-6032; Fax: 314-362-1461; e-mail: bgrossman@pathology.wustl.edu.
