
Abstract
Rare, congenital nonimmune hemolytic disorders of the erythrocyte, although uncommon, are important causes of anemia in the child and adult. These are a heterogeneous group of diseases that disrupt normal erythrocyte structure and function in varying ways. Predominant are abnormalities of hemoglobin stability, defects of erythrocyte metabolism, and disorders of erythrocyte hydration. Unstable hemoglobinopathies may lead to chronic or episodic hemolysis. Perturbation of critical enzymes of the Embden–Meyerhof pathway lead to altered erythrocyte metabolism and chronic hemolysis. Disorders of erythrocyte hydration are an under-recognized cause of hemolytic anemia. Beyond pathophysiologic mechanisms of disease, clinical, laboratory, and genetic heterogeneity characterize this group of disorders. Often, they are underdiagnosed or misdiagnosed. This review discusses pathophysiology, inheritance, clinical findings, laboratory manifestations, and management considerations in several rare nonimmune hemolytic diseases including the unstable hemoglobins, disorders of erythrocyte metabolism, and abnormalities of erythrocyte hydration.
Learning Objectives
Define important characteristics of 3 uncommon nonimmume hemolytic disorders, unstable hemoglobins, metabolic disorders of the erythrocyte, and abnormalities of erythrocyte dehydration
Specific strategies to allow recognition and diagnosis of these disorders are provided
Intrinsic disorders of the erythrocyte are a common cause of hemolytic anemia in children and adults. Typically, these disorders are divided into three categories, abnormalities of hemoglobin, defects of erythrocyte metabolism, and disorders of the erythrocyte membrane. The most common of these disorders are the alpha and beta hemoglobinopathies, glucose-6-phosphate dehydrogenase (G6PD) deficiency, and hereditary spherocytosis, affecting millions of people worldwide.
Rare intrinsic disorders of the erythrocyte that lead to hemolytic anemia can also be classified into these categories. Unstable hemoglobinopathies are a rare, often undiagnosed, important cause of hemolytic anemia. Disorders of the Embden–Meyerhof pathway may lead to altered erythrocyte metabolism. In both cases, symptomatology may be chronic or episodic and diagnosis may be delayed.1 Finally, disorders of membrane proteins leading to altered erythrocyte hydration are a frequently misdiagnosed or unrecognized cause of hemolytic anemia. This review discusses pathophysiology, inheritance, clinical findings, laboratory manifestations, and management considerations in several rare nonimmune hemolytic diseases including the unstable hemoglobins, disorders of erythrocyte metabolism, and abnormalities of erythrocyte hydration.
Unstable hemoglobins
Unstable hemoglobins result from globin chain mutations that alter the structure of the hemoglobin tetramer, leading to its instability, altered solubility, and intracellular precipitation. These globular, intracellular globin precipitates, called Heinz bodies, bind to the erythrocyte membrane decreasing membrane deformability and increasing membrane permeability. As a result, affected erythrocytes have a reduced lifespan. These disorders are sometimes called the congenital Heinz body hemolytic anemias.
Pathophysiology and inheritance
Mutations in any of the globin chains can led to unstable hemoglobins. Mutations altering the primary sequence of alpha-, beta-, gamma-, and delta-globin chains have been described. Several mechanisms may bring about altered globin chain solubility. These include mutations that weaken or alter heme–globin interactions preventing formation of the intact hemoglobin tetramer, mutations that interfere with the secondary or tertiary structure of hemoglobin subunits, or mutations that affect subunit interactions, such as interference with the quaternary structure of hemoglobin. Rare cases have been associated with mutations that alter interactions with alpha hemoglobin stabilizing protein.
Altered hemoglobin stability leads to its denaturation with the production of hemichromes and formation of Heinz bodies. Hemichromes bind to the membrane and decrease erythrocyte deformability, leading to splenic trapping and shortening erythrocyte lifespan.2,3 Oxidative damage from free heme iron may further compromise the membrane. Common in unstable hemoglobinopathies, but also found in some enzymopathies and thalassemia, Heinz bodies are intracellular precipitates seen after supravital staining as dark globular aggregates. Splenic pitting of Heinz body-containing erythrocytes is the process in which a section of the membrane and the Heinz body inclusion is removed from the erythrocyte during its passage through splenic sinusoids, forming a “bite” or “blister” cell (Figure 1). Erythrocyte membrane loss eventually leads to removal from circulation.4

Unstable hemoglobins: peripheral blood smear and Heinz body preparation. Peripheral smear (left) shows “bite” cells with pitted-out semicircular areas of the red blood cell membrane as a result of removal of Heinz bodies by macrophages in the spleen. The Heinz body preparation (middle) shows increased Heinz bodies in the same specimen compared with a control (right). Reproduced with permission from Benz and Ebert.41
Unstable hemoglobins: peripheral blood smear and Heinz body preparation. Peripheral smear (left) shows “bite” cells with pitted-out semicircular areas of the red blood cell membrane as a result of removal of Heinz bodies by macrophages in the spleen. The Heinz body preparation (middle) shows increased Heinz bodies in the same specimen compared with a control (right). Reproduced with permission from Benz and Ebert.41
Clinical severity is linked to the causative variant and its influence on hemoglobin stability. Another determinant of clinical severity is the influence of the mutation on oxygen affinity and thus its influence on the oxygen–hemoglobin dissociation curve. Unstable hemoglobin variants with high oxygen affinity tend to have milder anemia because of increased erythropoietin-stimulated erythropoiesis.
Inheritance is autosomal dominant in most cases. De novo mutations have been described, as have rare homozygous or compound heterozygous cases.
Hyperunstable hemoglobins are either barely detectable or undetectable in hemolysate. These rare hemoglobins are thought to be synthesized normally but due to their extreme instability are rapidly destroyed. This subgroup of unstable hemoglobins presents with a clinical phenotype similar to severe thalassemia, but with a dominant pattern of inheritance.
Clinical findings
Clinical presentation is variable. Patients typically present with chronic hemolytic anemia, ranging from asymptomatic to compensated hemolysis to severe anemia. Severely affected patients, particularly those with hyperunstable hemoglobins, present in infancy or early childhood and may require chronic transfusion therapy.
A hallmark of many of the unstable hemoglobinopathies is the propensity toward increased hemolysis when the patient experiences oxidant stress, such as exposure to sulfonamides, or during intercurrent infection. Some patients with unstable hemoglobins do not suffer chronic hemolysis, but only experience significant hemolysis after drug or other oxidant-induced stress.
Rare cases present in neonates with hemolytic anemia and jaundice that resolve in the first few months of life. These patients have unstable hemoglobinopathy due to mutations of the gamma-globin chain that resolves as fetal hemoglobin is replaced by adult.
An interesting finding in some cases of unstable hemoglobinopathy is the presence of dark urine or pigmenturia. This color change is due to the presence of dipyrrole methenes of the mesobilifuscin group. The origin of these fluorescent compounds is not clear. These methenes, 2 pyrrole rings linked by a methenyl bridge, have been linked to dysfunctional methenyl oxygenase, the enzyme involved in the breaking of −CH= bridges. Pigmenturia is not a sine qua non for the diagnosis of unstable hemoglobinopathy as fluorescent dipyrroles may also be found in Heinz bodies as part of formed hemichromes.
Laboratory manifestations and diagnosis
Patients exhibit minimal to mild to severe hemolytic anemia. Peripheral blood smear typically shows a normocytic anemia with nonspecific findings of hemolysis. In more severe cases, there is hypochromic anemia with decreased MCH due to removal of Heinz bodies and associated membrane loss. In severe cases, anisocytosis, basophilic stippling, Howell–Jolly bodies, microspherocytes, and occasional normoblasts may be seen. These findings are common in the hyperunstable hemoglobin variants. In some cases, elevated levels of methemoglobin may be detected.
Detection of Heinz bodies requires preincubation of erythrocytes with a supravital stain such as methylene blue. Heinz bodies are single or multiple small, <2μ inclusions, frequently associated with the erythrocyte membrane, (Figure 1) best detected after 24-48 hour incubation in the absence of glucose. Heinz bodies are easier to detect postsplenectomy and are rarely detected in infants <6 months of age. Because Heinz bodies are detected in unstable hemoglobinopathy, beta-thalassemia, and enzymopathies, their best use is as a screening test.
Hemoglobin electrophoresis or HPLC do not reliably detect all unstable hemoglobins, because many undergo rapid denaturation and degradation within the cell, leaving only normal hemoglobin visible on testing. Occasionally, the unstable hemoglobin appears as a diffuse, rather than a sharp, well-defined band, likely because of partial denaturation of hemoglobin during sample processing or electrophoresis. Sometimes, no bands are seen, but precipitates are seen at site of sample loading. Hemoglobin electrophoresis alone should not be relied on for diagnosis of unstable hemoglobins.
Examination of hemoglobin stability by heat or isopropanol is the most frequently used screening test in detection of these disorders. In alcohol precipitation testing, after isopropanol incubation, unstable hemoglobins yield visible precipitates. Similarly, in heat stability testing, hemolysate is incubated for 1-2 hours at 50°C. Unstable hemoglobins yield a visible precipitate while normal hemoglobins do not. Elevated HbF levels and methemoglobinemia can yield unreliable results in stability testing and results are not reliable in infants <6 months of age. Thus, the combination of hemoglobin electrophoresis and hemoglobin stability testing are frequently performed in the initial diagnostic evaluation.
DNA sequencing of the globin genes will determine the diagnosis in most cases, even cases of hyperunstable hemoglobins. In neonates and young infants, sequence of the gamma-globin chains should also be determined.
Treatment and outcome
The majority of unstable hemoglobinopathies are mild disorders and require only supportive care and expectant management.5,6 Patient education regarding prevention, ie, avoidance of oxidant drugs, and treatment, emphasizing prompt evaluation during hemolytic episodes, should be provided.
Transfusion may be required during episodes of hemolysis and on a chronic basis in patients with severe disease. Splenectomy has been utilized with some benefit in patients with severe disease. Thromboembolic events have been noted following splenectomy for unstable hemoglobin.
Disorders of erythrocyte metabolism
Disorders of erythrocyte metabolism have been included in the term congenital nonspherocytic hemolytic anemia (CNSHA), a classification that traditionally includes erythrocyte disorders not due to defects of hemoglobin, the membrane, immune-mediated disease, or other diseases such as paroxysmal nocturnal hemoglobinuria. CNSHA is a heterogeneous group of inherited disorders associated with various defects in erythrocyte metabolism, including enzymopathies of glucose, glutathione, and nucleotide metabolism (Figure 2). Hemolysis occurs due to enzyme or antioxidant deficiency or dysfunction (eg, abnormal substrate or cofactor binding), altered activation or inhibition characteristics, or decreased stability or specific activity.

Pathways of energy metabolism in the erythrocyte. Glucose 6-phosphate may be degraded anaerobically to lactate through the Embden–Meyerhof pathway, or oxidatively through the hexose monophosphate shunt. Pentose phosphates (R-5-P) can reenter anaerobic glycolysis as fructose 6-phosphate (F-6-P) and glyceraldehyde 3-phosphate (G-3-P) after conversion by enzymes of the terminal pentose phosphate pathway or as a product of adenosine or inosine degradation. 2,3-diphosphoglycerate (2,3-DPG) may be generated instead of adenosine triphosphate (ATP) through diversion of triose through the Rapoport–Luebering shunt. Glutathione may be synthesized directly from constituent amino acids; its cycling from oxidized (GSSG) to reduced forms (GSH) depends on reduced pyridine cofactor (NADPH) generation. ADP indicates adenosine diphosphate; DHAP, dihydroxyacetone phosphate; FDP, fructose 1,6-diphosphate; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; and PEP, polyestradiol phosphate. Reproduced with permission from Gallagher.42
Pathways of energy metabolism in the erythrocyte. Glucose 6-phosphate may be degraded anaerobically to lactate through the Embden–Meyerhof pathway, or oxidatively through the hexose monophosphate shunt. Pentose phosphates (R-5-P) can reenter anaerobic glycolysis as fructose 6-phosphate (F-6-P) and glyceraldehyde 3-phosphate (G-3-P) after conversion by enzymes of the terminal pentose phosphate pathway or as a product of adenosine or inosine degradation. 2,3-diphosphoglycerate (2,3-DPG) may be generated instead of adenosine triphosphate (ATP) through diversion of triose through the Rapoport–Luebering shunt. Glutathione may be synthesized directly from constituent amino acids; its cycling from oxidized (GSSG) to reduced forms (GSH) depends on reduced pyridine cofactor (NADPH) generation. ADP indicates adenosine diphosphate; DHAP, dihydroxyacetone phosphate; FDP, fructose 1,6-diphosphate; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; and PEP, polyestradiol phosphate. Reproduced with permission from Gallagher.42
Pathophysiology and inheritance
At steady state, erythrocytes perform their primary functions, gas transport and exchange, without a net change in energy state. However, erythrocytes require ATP production to support many cellular processes including membrane ion transport, maintenance of phospholipid asymmetry, glutathione synthesis, purine and pyrimidine metabolism, and maintenance of a reducing state to prevent widespread oxidant damage. Because erythrocytes do not undergo oxidative phosphorylation and cannot store glycogen, they need to continuously catabolize glucose from the blood, primarily from glycolysis (Embden–Meyerhof pathway, 90% steady-state ATP production) and the pentose phosphate pathway (hexose monophosphate shunt, 5%-10% steady-state ATP production) as sources of ATP (Figure 2).
Pyruvate kinase (PK) deficiency accounts for ∼90% of inherited defects of glycolysis.7 It is the second most common inherited erythrocyte enzymopathy after G6PD deficiency. PK deficiency is found worldwide, but it is most common in individuals of northern European and Asian descent.
Defects of glycolysis are inherited in an autosomal recessive manner.8 Unaffected heterozygous carriers are clinically normal, although their erythrocytes may contain decreased amounts of mutant enzyme. Phosphoglycerate kinase deficiency is an exception, as it is an X-linked disorder with hemolytic anemia typically found in males.
Clinical findings
Severity of hemolysis is variable in these disorders, ranging from asymptomatic, well compensated anemia to transfusion-dependent anemia (Table 1). Severely affected patients present in infancy or early childhood and rare cases have presented in utero with severe fetal anemia. Some patients with mild disease escape detection until later in life when complications related to chronic hemolysis occur, the patient develops an aplastic crisis, or when the diagnosis is made during evaluation for another condition. In most cases, hemolysis is chronic and it is not typically influenced by drugs or other inciting agents. Mutant erythrocytes with low levels of ATP are trapped in the hostile environment of the spleen, leading to shortened erythrocyte life span and splenomegaly.
Hemolytic anemia: associated symptoms and benefit of splenectomy in enzyme-linked hemolytic anemias
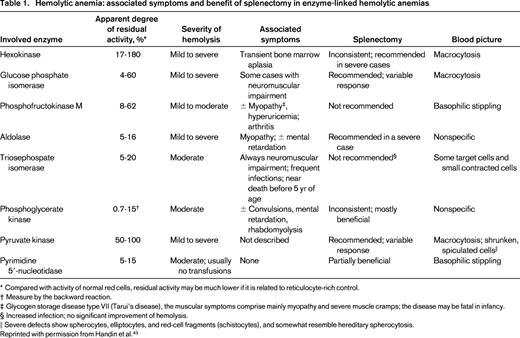
* Compared with activity of normal red cells, residual activity may be much lower if it is related to reticulocyte-rich control.
† Measure by the backward reaction.
‡ Glycogen storage disease type VII (Tarui's disease), the muscular symptoms comprise mainly myopathy and severe muscle cramps; the disease may be fatal in infancy.
§ Increased infection; no significant improvement of hemolysis.
‖ Severe defects show spherocytes, elliptocytes, and red-cell fragments (schistocytes), and somewhat resemble hereditary spherocytosis.
Reprinted with permission from Handin et al.43
PK deficiency typifies the heterogeneity of these disorders, ranging from in utero hemolysis and neonatal transfusion dependence to mild disease not detected until late in life.9 Because PK catalyzes the conversion of phosphoenolpyruvate to pyruvate to generate ATP, abnormalities or deficiency of PK leads to erythrocyte energy depletion with perturbation of many ATP-dependent cellular processes. Upstream catabolites accumulate in the red cell, including 2,3-DPG, which shifts the oxygen dissociation curve to the right, enhancing tissue oxygenation and ameliorating some of the physiologic effects of anemia. Iron overload sometimes occurs in PK deficiency even outside the stage of chronic transfusion or co-inherited hemachromatosis.10 Study of marrow or spleen from selected, iron-overloaded patients suggest there is significant ineffective erythropoiesis.2
Variable nonerythroid manifestations are found in many of the CNSHA disorders, primarily associated with muscle and/or nervous system involvement (Table 1). These disorders include glucose phosphate isomerase (GPI) deficiency, phosphofructokinase (PFK) deficiency, triosephosphate isomerase (TPI), and phosphoglycerate kinase (PGK) deficiency.
GPI deficiency usually presents in infancy or early childhood with moderate to severe hemolytic anemia.8,13 Rare cases may be complicated by irreversible neurologic manifestations including hypotonia, myopathy, ataxia, dysarthria, and developmental delay.
PFK deficiency may involve erythrocytes, muscle, or both. Presentation is usually in adolescence with exertional myopathy. Affected patients typically suffer from a well-compensated hemolytic anemia. In the erythrocyte, PFK is a tetrameric enzyme composed of liver (L) and muscle (M) isoforms. Because most mutations are in the M isoform, partial deficiency of PFK activity is observed in erythrocytes from formation of L type homotetramers.
TPI deficiency is a rare disorder characterized by hemolytic anemia, cardiomyopathy, susceptibility to infection and progressive neuromuscular impairment beginning in infancy.14 Findings include dystonia, tremor, dyskinesia, pyramidal tract signs, severe weakness, and muscle wasting.15 The high frequency of a common European allele associated with TPI deficiency is inconsistent with the rare diagnosis of TPI deficiency, suggesting that homozygosity for mutant alleles is frequently embryonic lethal. Frequent miscarriages in couples with affected children support this hypothesis.
PGK deficiency may present with central nervous system abnormalities, seizures, and developmental delay with concomitant CNSHA, isolated myopathy without CNSHA, and CNSHA without CNS involvement.16,17 Females heterozygous for PGK defects may exhibit anemia associated with variable degrees of enzyme activity.
Other rare inherited abnormalities of glycolysis have been described. Hexokinase deficiency exhibits significant clinical, laboratory, and genetic heterogeneity. The most severe cases present as neonates and require chronic transfusion therapy. Hemolytic anemia has also been described in isolated cases of 2,3-bisphosphoglycerate mutase deficiency.
Disorders of nucleotide metabolism.
The mature erythrocyte lacks the ability to synthesize purine and pyrimidine nucleotides de novo. However, some nucleotides are formed through salvage pathways. In these salvage pathways, pyrimidine 5′-nucleotidase (P5N) degrades the pyrimidine nucleotides of RNA to cytidine and uridine, which then diffuse out of the erythrocyte. In cases of P5N deficiency,18 nondiffusible, partially degraded RNAs accumulate, leading to basophilic stippling characteristic of P5N-deficient erythrocytes. These collected pyrimidine nucleotides inhibit the transport of GSSG out of red cells, leading to high levels of erythrocyte glutathione. Affected patients suffer from mild to moderate hemolytic anemia with splenomegaly. Splenectomy does not typically ameliorate the hemolysis.
Laboratory manifestations and diagnosis
Variable degrees of anemia are present. Peripheral blood smears, with the exception of P5N deficiency, are nondiagnostic. Extensive spiculation of erythrocytes, especially postsplenectomy, is claimed to be characteristic of pyruvate kinase deficiency.19 Although occasional contracted, dense, spiculated cells may sometimes be found on peripheral blood smears of a pyruvate kinase-deficient patients,20 they are nonspecific. In contrast, basophilic stippling of erythrocytes is characteristic of P5N deficiency.
Osmotic fragility of fresh erythrocytes is normal. A population of osmotically fragile cells after incubation may be found in some patients.
Enzyme assays.
Fluorescent-based screening tests have been developed for use in these disorders, but diagnosis is best made by qualitative or quantitative assays of specific enzyme activity. An integrated series of enzyme assays has been developed, allowing detection of enzyme deficiency in most cases.21 Measurement of glycolytic intermediates may assist in diagnosis, as concentrations of upstream intermediates are typically increased upstream of a defect and concentrations of downstream intermediates are decreased. In most cases, heterozygotes have intermediate enzyme levels.
Enzyme assays should be interpreted with caution: These studies only assay circulating erythrocytes and not cells already hemolyzed in vivo. Enzyme assay conditions in vitro may not accurately reflect the in vivo environment. Measurement of enzymes from transfused erythrocytes or contaminating leukocytes may obscure the underlying metabolic defect. Leukocyte depletion from samples is essential as they contain significant amounts of glycolytic enzymes. Finally, averages of enzyme activity may not accurately reflect activity in subpopulations of erythrocytes, particularly aged cells. This is particularly true when there is reticulocytosis, when younger cells with higher enzyme levels yield artificially elevated mean levels.
The diagnosis of P5N deficiency can be suspected by making use of the difference between the ultraviolet absorption spectrum of pyrimidine and purine nucleotides.23 Enzyme testing can confirm enzyme deficiency.
DNA analysis.
Application of molecular genetic analyses including DNA sequencing and other molecular studies complement enzyme assays and provide definitive diagnosis in most cases. Sequencing of individual exons and adjacent intron sequences and promoters of candidate genes, available in several commercial laboratories, is the initial step.7,8,22 Because the majority of metabolic disorders are inherited in a recessive manner, identification of 2 heterozygous mutations mandates the parents are studied to ensure the mutations are in trans. Alternatively, mRNA can be isolated from reticulocytes, reverse transcribed, amplified, and amplification products subcloned and sequenced to ascertain whether the mutations are in cis or in trans. In some disorders, particularly PK deficiency, intragenic deletions missed by sequencing may be the underlying pathogenetic defect. Detection by MLPA, gap PCR, or other methodology is necessary for molecular diagnosis in these cases. Molecular analyses also have potential pitfalls. In some cases, variants of unknown significance are detected, making genetic diagnosis uncertain. Other mutations not detected by these techniques may be causative, eg, in distant regulatory elements, deep intronic splicing mutations, etc. In these cases, diagnosis is assigned based on clinical, laboratory, and biochemical findings.
Thus in many cases, particularly difficult ones, ones where parents are unavailable for study, etc, enzyme assays and molecular studies are complementary techniques.
Treatment and outcome
Most patients with CNSHA require only supportive care and expectant management similar to any patient with chronic hemolytic anemia. Transfusions are rarely needed except during aplastic episodes, during severe intercurrent illnesses, etc.
In severely affected patients, especially transfusion-dependent patient, splenectomy may lessen hemolysis and ameliorate anemia. Some patients, particularly PK-deficient patients, develop marked reticulocytosis postspelenectomy, up to 50%-70%. This paradoxical reticulocytosis has been attributed to increased reticulocyte survival after removal of the hostile splenic environment and alterations in the maturation of erythroid splenic progenitors.
In severely affected patients who do not respond to splenectomy, hematopoietic stem cell transplantation may be considered.
Patients requiring frequent transfusions and some nontransfused PK-deficient patients with ineffective erythropoiesis or coinherited hemochromatosis may require chelation therapy.
Disorders of erythrocyte hydration
Disorders of erythrocyte hydration are a heterogeneous group of disorders associated with abnormalities of erythrocyte solute content. Depending on the severity of the abnormalities of water and solute content and their effect on erythrocyte volume homeostasis, hemolytic anemia of varying severity may occur.24 These disorders have been classified as primary, due to intrinsic defects of erythrocyte hydration, and secondary, due to other intrinsic defects of the erythrocyte that also influence erythrocyte hydration such as dehydrated erythrocytes in sickle cell disease.25 Primary disorders of erythrocyte hydration include the hereditary xerocytosis syndromes with a dehydrated phenotype, the hereditary hydrocytosis syndromes with an overhydrated phenotype, and intermediate syndromes.
Hereditary xerocytosis
Pathophysiology and inheritance
The hereditary xerocytosis (HX) syndromes are the most commonly encountered disorders of erythrocyte hydration. The primary defect in HX erythrocytes is the passive loss of intracellular potassium in excess of accumulation of intracellular sodium, leading to a gradual decrease in total intracellular cations accompanied by obligate water loss to maintain osmotic balance.26 The HX erythrocyte is rigid because of its increased cytoplasmic viscosity and likely due to an intrinsic membrane rigidity induced by complexing of globin to spectrin. This rigidity and decreased deformability contribute to its shortened life span.
Missense mutations in PIEZO1 have been identified in most HX patients, primarily in the conserved COOH-terminal region (Figure 3).27-29 Piezo proteins are ion channels mediating mechanosensory transduction in mammalian cells.30 Animal models suggest mechanically activated Piezo1 plays a critical role in erythrocyte volume homeostasis.31 In vitro studies of HX-associated PIEZO1 mutations demonstrate a gain-of-function phenotype, with many mutants demonstrating delayed channel inactivation,28,32 suggesting increased cation permeability leads to HX erythrocyte dehydration. In a subset of HX patients, defects of the Gardos channel have been described.33-35 Inheritance is autosomal dominant.

A model of human PIEZO1 and associated mutations in hereditary xerocytosis. The location of PIEZO1 mutations identified in hereditary xerocytosis are shown. Reproduced with permission from Bagriantsev et al.30
A model of human PIEZO1 and associated mutations in hereditary xerocytosis. The location of PIEZO1 mutations identified in hereditary xerocytosis are shown. Reproduced with permission from Bagriantsev et al.30
Clinical features
The HX syndromes are characterized by mild to moderate, well compensated hemolytic anemia. Splenomegaly and other markers of chronic hemolysis are uncommon. Some patients do not come to medical attention until late in life. The HX syndromes are pleiotropic. Some patients suffer from transient perinatal edema and nonimmune hydrops fetalis that spontaneously resolve. Some HX patients suffer from pseudohyperkalemia.27,36
Iron overload is a significant finding in many adult HX patients, severe enough to require chelation, outside the setting of chronic transfusion.37 The basis for the iron overload is unknown.
Laboratory manifestations and diagnosis
Beyond anemia, the reticulocyte count in HX patients is often increased to a level higher than that expected for the degree of anemia. Erythrocyte morphology on peripheral blood smear is relatively normal, with a few target cells, variable numbers of stomatocytes and dessicytes-contracted erythrocytes with their hemoglobin pooled to the side (Figure 4). The dehydration of HX erythrocytes is reflected by elevated MCHC and decreased osmotic fragility curve (increased osmotic resistance). These tests allow differentiation of HX from hereditary spherocytosis, where the MCHC is elevated, but osmotic fragility is usually increased. Despite cellular dehydration, HX patients exhibit mildly elevated MCV. This is attributed in part to an artifact of cellular stiffness. In electronic cell counters, the conversion of pulse height to cellular volume is dependent on cell shape. HX erythrocytes do not deform to the same degree as normal erythrocytes, causing the electronically measured MCV to be estimated ∼10% too high. Reticulocytosis also contributes to the elevated MCV.

Peripheral blood smears in altered volume homeostasis. (Left) Hereditary xerocytosis. (Right) Hereditary hydrocytosis. (Left) A Wright-stained peripheral blood smear from a patient with hereditary xerocytosis due to a PIEZO1 mutation showing rare stomatocytes, occasional dessicytes–dense, abnormal erythrocyte forms where hemoglobin appears puddled at the periphery, and rare target cells. (Right) A Wright-stained peripheral blood smear from a patient with hereditary hydrocytosis is shown. Numerous stomatocytes, erythrocytes with a central mouth-like “stoma” are seen.
Peripheral blood smears in altered volume homeostasis. (Left) Hereditary xerocytosis. (Right) Hereditary hydrocytosis. (Left) A Wright-stained peripheral blood smear from a patient with hereditary xerocytosis due to a PIEZO1 mutation showing rare stomatocytes, occasional dessicytes–dense, abnormal erythrocyte forms where hemoglobin appears puddled at the periphery, and rare target cells. (Right) A Wright-stained peripheral blood smear from a patient with hereditary hydrocytosis is shown. Numerous stomatocytes, erythrocytes with a central mouth-like “stoma” are seen.
Specialized laboratory testing reveals reduced erythrocyte potassium concentration and total monovalent cation content. Osmotic gradient ektacytometry also has characteristic findings.38 DNA sequencing of the PIEZO1 or KCNN4 genes may demonstrate the causative mutation.
Treatment and outcome
No specific treatment is available or usually required. Splenectomy is of little or no benefit, and is considered contraindicated as it has predisposed to life-threatening thrombotic episodes in some HX patients.39 Complications of chronic hemolysis and erythropoietic stress may occur.
Hereditary hydrocytosis
The hereditary hydrocytosis syndromes, also known as hereditary stomatocytosis, are associated with elevated erythrocyte sodium concentration, reduced potassium concentration, and increased total sodium and potassium content.40 Excess cations elevate cell water leading to swollen, macrocytic, 110-150 fL, osmotically fragile erythrocytes with a low MCHC (24%-30%). Peripheral blood smear shows stomatocytosis (Figure 4). Missense mutations in the Rh-associated glycoprotein have been identified in a subset of hydrocytosis patients.26 Like the HX syndromes, splenectomy may be deleterious.39
Intermediate syndromes
HX and hereditary hydrocytosis represent the extremes of a spectrum of defects in erythrocyte hydration.24 Kindreds with features of both conditions have been reported. Interestingly, some patients with significant defects in hydration have little or no hemolysis. In these cases, numbers of stomatocytes and the degree of sodium influx do not correlate, and neither correlates with severity of hemolysis.
Acknowledgment
This work was supported by the Doris Duke Foundation and NIH Grant DK104046.
Correspondence
Patrick G. Gallagher, Department of Pediatrics, Yale University School of Medicine, 333 Cedar St, PO Box 208064, New Haven, CT 06520-8064; Phone: 203-688-2896; Fax: 203-785-6974; e-mail: patrick.gallagher@yale.edu.
