
Abstract
Hemolytic anemia (HA) is a frequent condition with variable pathophysiology. Hematopoietic stem cell transplantation (HSCT) is unique because it is performed across the ABO blood group barrier. Thereby, there is a transfer of plasma, red blood cells, and immunocompetent cells from the donor to the recipient, possibly leading to HA, due to red blood cell incompatibility. The underlying disease, drugs (particularly those used for conditioning and immunosuppressants), infections, graft-versus-host disease, and autoimmune diseases may all contribute to the clinical and laboratory picture of HA. Additionally, transplantation-associated thrombotic microangiopathy (TA-TMA) may occur and is associated with significant morbidity and mortality. This review highlights the current knowledge on HA after allogeneic HSCT, particularly due to ABO incompatibility. We follow the timeline of the transplantation process and discuss investigations, differential diagnosis, and prophylactic measures including graft processing to avoid hemolysis in case of ABO incompatibility. Finally, current therapeutic approaches for both TA-TMA and post-HSCT autoimmune HA, which are associated with high morbidity and mortality, are discussed.
Learning Objectives
To understand that hemolytic anemia (HA) is frequent after hematopoietic stem cell transplantation (HSCT)
To discuss different etiologies of HA during and after allogeneic HSCT
To know how to approach and investigate HA in this situation for an accurate diagnosis
To know the prophylactic measures to reduce the extent of hemolysis in case of ABO-incompatible HSCT and to know currently available therapeutic options
To know the special transfusion requirements of patients before, during, and after HSCT, implying a close collaboration between clinicians, transplant physicians, and transfusion services
Hemolytic anemia (HA) is a condition in which the patient's red blood cells (RBCs) are prematurely destroyed. HA in general is either inherited or acquired, intravascular or extravascular, and immune or nonimmune mediated. Hemolysis ranges from being asymptomatic and harmless to therapy resistant, life threatening, and even fatal.
Hematopoietic stem cell transplantation (HSCT) is a potentially curative and increasingly used treatment approach for different malignant and nonmalignant diseases, including entities associated with HA, such as chronic lymphocytic leukemia with autoimmune HA (AIHA), paroxysmal nocturnal hemoglobinuria, and sickle cell disease.1 HA can develop after HSCT; however, HSCT can still be considered for the treatment of severe, therapy-resistant AIHA.
Hemolysis during and after HSCT can occur at different time points, ie, even weeks or months after transplantation, and may have several causes (Figure 1). Investigation may be difficult because the differential diagnosis is often broad. As opposed to other reviews of HAs, most often structured according to the pathophysiology of the hemolysis (ie, immune vs nonimmune), in this review, we have followed the timeline of the transplantation process and have discussed the investigation, differential diagnosis, and management at the time points during transplantation when HA most commonly occur. Therefore, discussion of immune and nonimmune causes of hemolysis follows the chronological order of transplantation, and management of blood group incompatibility is discussed before transplantation-associated thrombotic microangiopathy (TA-TMA) and this before post-transplant AIHA. We have maintained this order throughout the review, the tables, and the graphical representation.

Hemolytic anemia conditions encountered before, during and after hematopoietic stem cell transplantation (HSCT). *All RBC concentrates should be γ-irradiated (25-30 Gy) and leukocyte reduced. Consider HLA-alloimmunization. ¶See Table 3. AH indicates acute hemolysis; AIHA, autoimmune hemolytic anemia; BM, bone marrow; CB, cord blood; CBC, complete blood count; CLL, chronic lymphocytic leukemia; CVID, common variable immunodeficiency; D, donor; DAT, direct antiglobulin test; DIC, diffuse intravascular coagulation; DIHA, drug-induced HA; LDH, lactate dehydrogenase; PBSC, periphereal blood stem cells; PLS, passenger lymphocyte syndrome; Plt, platelets; PNH, paroxysmal nocturnal hemoglobinuria; PRCA, pure red cell aplasia; PTLD, post-transplant lymphoproliferative disease; R, recipient; Rc, reticulocytes; SAA, severe aplastic anemia; and TMA, thrombotic microangiopathy.
Hemolytic anemia conditions encountered before, during and after hematopoietic stem cell transplantation (HSCT). *All RBC concentrates should be γ-irradiated (25-30 Gy) and leukocyte reduced. Consider HLA-alloimmunization. ¶See Table 3. AH indicates acute hemolysis; AIHA, autoimmune hemolytic anemia; BM, bone marrow; CB, cord blood; CBC, complete blood count; CLL, chronic lymphocytic leukemia; CVID, common variable immunodeficiency; D, donor; DAT, direct antiglobulin test; DIC, diffuse intravascular coagulation; DIHA, drug-induced HA; LDH, lactate dehydrogenase; PBSC, periphereal blood stem cells; PLS, passenger lymphocyte syndrome; Plt, platelets; PNH, paroxysmal nocturnal hemoglobinuria; PRCA, pure red cell aplasia; PTLD, post-transplant lymphoproliferative disease; R, recipient; Rc, reticulocytes; SAA, severe aplastic anemia; and TMA, thrombotic microangiopathy.
HA in association with the underlying disease and infection-associated HA are beyond the scope of this review and will not be further discussed. However, they are listed in Table 1. We also refer to other sources.2-4 Drug-induced HA should always be considered, especially due to antimicrobial agents (eg, dapsone, penicillins, and cephalosporins) and immunosuppressants [calcineurin-inhibitors and sirolimus, which are the most frequently used drugs for graft-versus-host disease (GVHD) prophylaxis].5 Hemolysis due to passive transfer of antibodies from a high-titer type O blood product and hemolytic transfusion reactions (acute and delayed) following transfusion errors or due to non-ABO-RBC alloantibodies need to be excluded. In addition, every HSCT candidate, as well as the corresponding donor, can have additional conditions leading to HA (eg, glucose-6-phosphate dehydrogenase deficiency). Finally, disease relapse needs to be considered and ruled out.
Hemolytic conditions in allogeneic hematopoietic stem cell transplant recipients
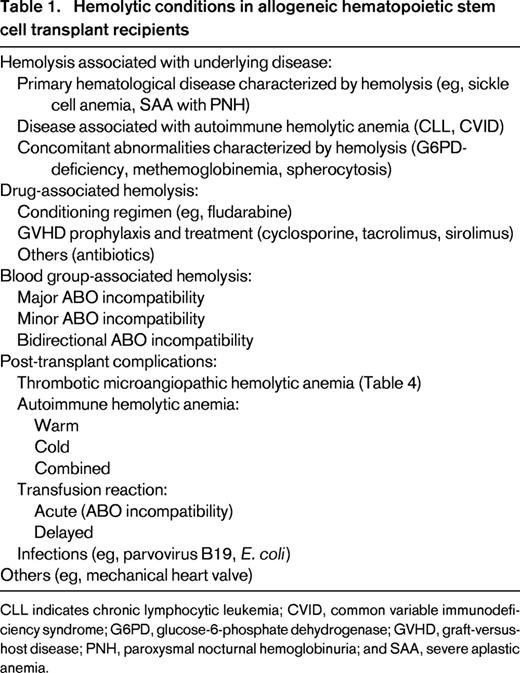
CLL indicates chronic lymphocytic leukemia; CVID, common variable immunodeficiency syndrome; G6PD, glucose-6-phosphate dehydrogenase; GVHD, graft-versus-host disease; PNH, paroxysmal nocturnal hemoglobinuria; and SAA, severe aplastic anemia.
Optimal management of HA after allogeneic HSCT implies an interdisciplinary approach and a close collaboration between clinicians, transfusion service and blood bank and the stem cell laboratory. Because supportive care with transfusions constitutes an important component of the management of HA in this setting, special attention has to be paid to transfusion practices.6 In general, all RBC concentrates should be γ-irradiated (25-30 Gy) and leukocyte reduced in order to reduce almost always fatal transfusion-associated GVHD and other transfusion reactions.
Diagnostic workup
A stepwise diagnostic workup with reasonable investigations is the basis for an accurate diagnosis and appropriate therapy. The patient's history, knowledge of the performed transplant procedure (type and intensity of conditioning, donor and recipient ABO blood group, graft source, and GVHD prophylaxis and therapy) and the patient's transfusion history are essential. Hemoglobin monitoring (sometimes repetitively in 1 day in case of severe hemolysis), a full blood count including reticulocytes, blood smear (schistocytes? microspherocytes?), and blood chemistry [bilirubin, lactate dehydrogenase (LDH), and creatinine] are mandatory. The evaluation of haptoglobin and free hemoglobin in serum and urine can be helpful. In case of immune-mediated hemolysis, a direct antiglobulin test (DAT), elution (also against a non-O RBC panel in case of ABO incompatibility), isohemagglutinin titration, and absorption techniques are required. In the presence of schistocytes and thus the suspicion of microangiopathy, measurement of ADAMTS13 should be considered. In addition, acute and delayed transfusion reactions because of a transfusion error should always be excluded, according to the local policies.
Prevention and management of HA due to blood group incompatibility
In contrast to solid organ transplantation, donor-recipient ABO incompatibility is not an impediment for HSCT and occurs in ∼30%-50% of transplants.7,8 In major ABO-incompatible HSCT, the patient has preformed antibodies (ie, isohemagglutinins) against A and/or B antigens expressed on the donor's RBC. Minor ABO-incompatible HSCT is characterized by the transfer of donor isohemagglutinins directed against the recipient's RBC antigens. A bidirectional blood-group barrier is a combination of major and minor ABO incompatibilities. The presence of these isohemagglutinins and the involvement of the donor's and recipient's immune system are responsible for hemolytic complications (Table 2). Special attention should thus be paid to the donor's ABO blood group and the stem cell source, because they differ in terms of the volume of RBC and plasma, and number of lymphocytes.9 RBC antigens are also expressed on other tissues, including endothelial cells (“histo-blood groups”). Therefore, one may speculate that ABO incompatibility could have an association with the pathogenesis of GVHD. However, many studies show discrepant results regarding transplant outcomes and it is most likely that ABO blood-group incompatibility is not important for transplant outcome.7,8
Hemolytic complications due to ABO incompatibility

Bidirectional ABO incompatibility: combination of both major and minor ABO incompatibilities.
ATG indicates anti-thymocyte globulin; DLI, donor-lymphocyte infusion; EPO, erythropoietin; PLS, passenger lymphocyte syndrome; RBC, red blood cell; and TPE, plasma exchange.
Major ABO incompatibility
Acute HA can occur during and immediately after graft infusion as a consequence of donor's RBC hemolysis. This varies depending on the graft source, as bone marrow contains more RBCs compared with peripheral blood progenitor cells (PBSCs) collected by apheresis and cord blood (CB).
Donor's RBCs can be depleted from the graft through different graft processing steps (apheresis or sedimentation) at the expense of a loss of viable progenitor cells.8,10 Red cell reduction should be performed targeting a packed red cell content <20-25 mL.11 On the other hand, acute hemolysis can be prevented or at least tempered through reduction of recipient's isohemagglutinin titers through infusion of secretor plasma, therapeutic plasma exchange (TPE), or immunoadsorption.12 Some centers transfuse before HSCT donor-type, incompatible RBCs with consequent in vivo adsorption limited to patients receiving myeloablative conditioning.13 In case of in vivo adsorption, patients have to be closely monitored for acute hemolytic transfusion reactions and adequately hydrated to preserve renal function. TPE and immunoadsorption have to be performed before major ABO-incompatible HSCT on a daily basis with the goal to reduce the IgM and/or IgG antibody titers. However, clinicians should be aware that titer determination is not standardized and shows a wide intra-individual variability. Additionally, IgM isohemagglutinins are removed more efficiently than IgG isohemagglutinins, because IgG distributes in both the intravascular and extravascular spaces.14 Furthermore, no consensus on target titer values is available. Thus, clinical relevant and serious acute hemolytic reactions immediately after graft infusion are rare. Nevertheless, major ABO-incompatibility needs to be considered and appropriately ruled out in case of acute reactions after transplantation.
Delayed red cell engraftment due to host anti-donor isohemagglutinins may occur. Pure red cell aplasia (PRCA) may develop in up to 30% of patients after major ABO-incompatible HSCT, because of persistence of recipient plasma cells producing anti-donor isohemagglutinins, thus blocking normal erythroid maturation.8,15 Delayed red cell engraftment and PRCA are more common in reduced intensity transplantation (RIC) where donor and recipient hematopoiesis coexist and in cord blood transplantation. After RIC there is longer persistence of recipient isohemagglutinins producing plasma cells than after myeloablative conditioning. Anemia, reticulocytopenia, and a bone marrow lacking erythroid precursors are clues for the diagnosis of PRCA in the setting of major ABO-incompatible HSCT. Parvovirus B19 infection has to be excluded. For this purpose, specific polymerase chain reaction from bone marrow specimens is considered to be a standard. As long as PRCA persists (usually weeks to months until the isohemagglutinins have been adsorbed), transfusion requirements are high with consequent iron overload and potentially negative impact on overall survival.16 Management of post-transplant PRCA may include (besides transfusions) rituximab, anti-thymocyte globulin, TPE or immunoadsorption, decrease/discontinuation of immunosuppression, and donor lymphocyte infusions.8 Although plausible from a pathophysiologic point of view, none of these practices have been proven to be effective.
Minor ABO incompatibility
Acute hemolysis may also rarely occur after minor ABO-incompatible HSCT through transfer of high-titer donor isohemagglutinins contained in the graft or in recipients with small blood volume (pediatric patients). This can be prevented through plasma volume reduction of the product.17
Passenger lymphocyte syndrome (PLS) is a significant and unpredictable complication after minor ABO-incompatible HSCT.18 It usually occurs 1-3 weeks after HSCT and is due to hemolysis of recipient's RBCs through isohemagglutinins produced by donor-derived immunocompetent lymphocytes. Hemolysis can be severe, even fatal, and persists until all the recipient RBCs are replaced by transfused or donor-derived RBCs. PLS is more common in patients with blood group A, with a donor of group O, and cyclosporine A (CYA) alone as GVHD prophylaxis. Monitoring for clinical and laboratory signs of hemolysis is mandatory and in case of massive hemolysis frequent hemoglobin measurements should be performed. DAT should be performed, although it can be negative in case of rapid clearance of isohemagglutinin-loaded recipient RBCs. In case of a positive DAT, elution against group A and/or B reagent RBCs (instead of the usual O group panel) can be helpful to support the diagnosis. Sometimes, isohemagglutinins against recipient ABO blood group antigens can be detected. Management consists primarily of adequate supportive care with transfusions of RBCs compatible with both the recipient and the donor. In some selected cases, RBC exchange can be performed.14
Bidirectional ABO incompatibility
This is defined as a combination of both major and minor ABO incompatibilities along with the risk of their consequences, and thus clinicians have to be aware of all the above-described complications.
In summary, awareness of possible complications after ABO-incompatible HSCT and early recognition and institution of appropriate measures are essential. Additionally, each center should define policies and standard operating procedures for the prevention and management of complications after ABO-incompatible HSCT (Table 3).19 Definite ABO blood group assignment should be done after a transfusion-independent interval, full engraftment, remission of the underlying disease, and in close collaboration with the treating physicians.

HA due to antibodies against non-ABO RBC antigens
Due to the multitude of RBC antigens, it is impossible to match stem cell donors, blood donors, and recipients for all these antigens. In contrast to ABO incompatibility, donors and recipients lack naturally formed antibodies for non-ABO RBC antigens, occurring only after immunization. Donors are screened for alloantibodies. Unrelated donors in general have no history of transfusions; in related donors, where donor eligibility is less rigorous, careful transfusion and exposure history are important. Importantly, alloantibodies can occur against antigens of donor, recipient, and third party-transfused RBCs. Therefore, HA can also occur as a consequence of alloantibodies against non-ABO RBC antigens and has the same pathophysiology as PLS.8,20,21 The Rhesus (Rh) system is the one most frequently described. In case of preformed alloantibodies (through transfusions or pregnancy) and a major RhD incompatibility, delayed HA may result. In the case of minor incompatibility both immediate and delayed hemolysis can occur.21 In this case, management is similar to ABO-incompatibility. Transfusion support consists primarily in transfusion of RBC concentrates lacking the corresponding antigen. Additionally, RhD alloimmunization through platelet transfusions should be prevented either by choosing platelet concentrates from RhD-negative donors or through prophylaxis with anti-RhD immunoglobulins.
ABO incompatibility and platelet/plasma transfusion support
ABO-incompatible platelet transfusions can cause hemolysis, in particular, platelet concentrates from donors with high isohemagglutinin titers. Some transfusion services measure anti-A and/or anti-B titers, and thus units with high titers of isohemagglutinins can be transfused to ABO-identical recipients. However, there is no accepted and clear definition for high-titer antibodies. Platelets in additive solutions contain less donor plasma and thus less isohemagglutinins, and should therefore be preferred to standard plasma-suspended platelets. Only in rare cases, platelet components have to be washed.
Furthermore, transfusion of incompatible plasma is associated with increased transplant-related mortality due to an increased risk of infection, veno-occlusive disease, and multi-organ failure.22,23 Therefore, both donor- and recipient-compatible plasma should be transfused after HSCT to avoid hemolysis, due to the passive transfer of isohemagglutinins against recipient and/or donor RBC antigens (Table 3). AB plasma is the universal donor source.
HA can also occur after high doses of intravenous immunoglobulins (IVIGs), as these products are manufactured from human plasma and some of them may contain isohemagglutinins if the manufacturing process does not include a removal step.24 IVIGs are often administered to patients after HSCT to prevent or treat infectious complications. IVIG formulations with low isohemagglutinin titers and/or adjustment of dosage can prevent IVIG-induced HA, especially for patients with blood group A.
Thrombotic microangiopathic HA after HSCT

TA-TMA
TMA is a well-recognized complication after HSCT (TA-TMA). Frequency varies according to reports and may be seen in up to 35% of patients, depending on the diagnostic criteria and definitions.26-28 In contrast to thrombotic thrombocytopenic purpura (TTP), where an inborn or acquired deficiency of the von Willebrand factor multimer cleaving protease ADAMTS13 is the cause, the exact etiology and pathophysiology of TA-TMA remain unclear.25,28-30 Clinical presentation is heterogeneous and it is likely that TA-TMA represents a clinical syndrome that is a common end product of different pathophysiologic processes involving also the coagulation system. Risk factors, including endothelial damage by conditioning agents (including irradiation), medications (immunosuppressants like calcineurin inhibitors and sirolimus), and viral infections have been identified. There is an association between TA-TMA and GVHD, although causality remains to be proven. Complement system abnormalities including regulatory defects and autoantibodies against factor H have been described, which suggests a possible role of complement in the disease process. Latter is also supported by growing data on the use of eculizumab in TA-TMA.28-33
A high index of suspicion is required for the diagnosis of TA-TMA. Microangiopathic HA is characterized by the presence of anemia, low platelets, and schistocytes in a blood smear. Serum creatinine, LDH, bilirubin, and serum/urine-free hemoglobin (compatible with intravascular hemolysis) can be elevated; haptoglobin is usually decreased. In addition, hypertension and proteinuria can be the early signs of TA-TMA, although these manifestations are encountered frequently in patients after HSCT.26,27,34,35 Soluble membrane attack complex (sC5b-9) may be elevated and is associated with a poor prognosis.30 Diagnosis can be confirmed by renal biopsy, which shows typical histologic findings, although there is little correlation between clinical and pathologic diagnosis. However, this is rarely done and potential bleeding risks have to be balanced against the diagnostic benefits of this procedure.28 Unfortunately, there are no controlled trials and thus there is no consensus on the management of TA-TMA. Primarily, calcineurin inhibitors and/or sirolimus should be reduced in dose or discontinued if alternative drugs for the prevention or treatment of GVHD can be administered (eg, steroids, mycophenolate mofetil). This has to be balanced against the potential risk of GVHD. To which extent the above-mentioned immunosuppressants are directly responsible for or sustain TA-TMA remains speculative. Plasma infusion and TPE, based on their effectiveness in TTP, have not been proven to be effective, and controlled studies are lacking.14 Therefore, in the absence of enough evidence, we do not suggest TPE for the treatment of TA-TMA, even if some authors suggest an early initiation of daily TPE.36 Single case reports and case series have shown some success of rituximab, defibrotide, vincristine, and pravastatin.29,36 Complement blockade with eculizumab seems to be promising in patients with TA-TMA, although larger prospective studies are needed.30,37 Treatment remains overall unsatisfactory and morbidity and mortality in patients with TA-TMA are high, primarily due to renal impairment.38
Drug-induced TMA
Different drugs can cause TMA, through an immunologic reaction or because of direct toxicity, although the exact mechanism remains unclear.25 A recent systematic review supported a definite association of TMA with CYA, tacrolimus, and sirolimus, which are the immunosuppressants most commonly used for prophylaxis and treatment of acute and chronic GVHD.39-41 It is believed that these drugs exert a direct toxic effect, which can be dependent on dose or duration. In general, switching to another calcineurin inhibitor or sirolimus is not recommended. Other etiologies of TMA should be excluded, although the discrimination between drug-induced TMA and TA-TMA in transplanted patients is difficult. Additionally, differential diagnosis is not always obvious and patients can present with several potential risk factors for TMA (Table 4). All other drugs have to be critically reviewed and withdrawn if appropriate. Again, evidence is too weak to support treatment with TPE.14,41
Autoimmune HA
Autoimmune diseases (ADs) after both autologous and allogeneic (including cord blood) HSCT may occur regardless of the underlying disease.42-44 The exact mechanisms and the pathophysiology of post-transplant ADs are not yet fully understood. Failure of central and/or peripheral tolerance is believed to be involved in the escape of auto-reactive lymphocytes, thus leading, if uncontrolled, to the development of ADs. Infections, which occur frequently in HSCT recipients as a consequence of their disease, conditioning, and immunosuppression, may play an additional role in the pathogenesis of post-transplant ADs.42
In general, AD can affect every organ and occur alone or in combination.42 Autoimmune cytopenias after HSCT (including AIHA, immune thrombocytopenia, and immune neutropenia, or a combination of them) occur frequently.45-47 Incidence ranges from 1.3% to 4.4% and the risk factors for the development of AIHA are transplantation from an unrelated donor, development of chronic GVHD and a nonmalignant primary disease.45 Disease course is variable, ranging from spontaneous remissions to life-threatening and even fatal hemolysis. Post-transplant AIHA is often therapy resistant and associated with decreased survival.
Diagnosis of post-transplant AIHA has to be distinguished from disease relapse, graft failure, drug- and treatment-related toxicity, infection, and GVHD. Patients have clinical and laboratory evidence of HA, a positive DAT (usually positive for IgG ± C3d in warm-type and positive for C3d in cold-type AIHA), and a positive, panreactive indirect antiglobulin test. To exclude any underlying alloantibody, which carries the risk of delayed hemolytic transfusion reactions, time-consuming absorption techniques and/or knowledge of blood-group genotype are needed. Other causes of HA should be excluded. Evidence for treatment of post-transplant AIHA is lacking and available data arise from single case reports or case series. Steroids should be administered at a dosage of 1-2 mg/kg. In refractory patients, rituximab and other immunosuppressive drugs including combinations can be added.45,47 Immunosuppression has to be balanced against the risks of disease relapse and infections. Splenectomy can be recommended to patients without contraindications. In cold-type AIHA, avoidance of cold exposure is essential, as immunosuppression is less effective. If blood transfusions are indicated, crossmatching can be unable to identify compatible RBC units, as the autoantibodies are directed against highly prevalent antigens. However, transfusion requirement in acute AIHA can be a medical emergency and must not be delayed as RBC transfusions can be lifesaving.
Other HAs after allogeneic stem cell transplantation
Various malignant and nonmalignant diseases are associated with immune-mediated or nonimmune hemolysis. These diseases may relapse and thus HA can be a possible clinical manifestation either of relapse or of graft failure. In case of relapse, isohemagglutinins produced from surviving recipient plasma cells can drive HA through destruction of donor RBCs. In addition, due to immunosuppression, patients are at a risk of various infections, which in turn can cause HA or result in the development of post-transplant lymphoproliferative diseases; the latter, in rare cases, can manifest as AIHA.48
Future directions
Prospects through stem cell manipulation and graft processing have to be followed in the future. Importantly, a higher degree of standardization in the field of graft processing is needed. Further studies to better understand the pathophysiology of TA-TMA are needed. Moreover, new drug developments for prophylaxis and therapy of GVHD will perhaps avoid drug-induced TMA. Finally, the risk factors for post-transplant AIHA should be better addressed and prospective studies on therapeutic options for this treatment-resistant complication are warranted.
Acknowledgments
We thank Andreas Buser and Jörg Halter for critically reviewing the manuscript.
Correspondence
Andreas Holbro, Division of Hematology, Department of Internal Medicine, University Hospital, Petersgraben 4, 4031 Basel, Switzerland; Phone: 0041-61-265-25-25; Fax: 0041-61-265-44-50; e-mail: andreas.holbro@usb.ch.
