
Abstract
The myelodysplastic/myeloproliferative neoplasms (MDS/MPNs) lie at the interphase of phenotypically opposing bone marrow malignancies. They are characterized by concomitant features of bone marrow failure and myeloproliferation and are generally associated with a poor prognosis. Although much is unknown with respect to the clinical course and molecular biology of MDS/MPNs, emerging research is beginning to uncover the key defining characteristics of this designation. In this review, we will discuss the features of MDS/MPN diseases that unify there clinical and molecular course and those that define distinct disease entities. We will discuss advances in genetics and MDS/MPN modeling, as well as translational discoveries that are anticipated to inform the diagnosis, prognostication, and treatment of MDS/MPNs in the near future.
The myelodysplastic/myeloproliferative neoplasms (MDS/MPNs) are a unique group of myeloid malignancies that converge on a paradoxical bone marrow phenotype hallmarked by myeloid lineage subset proliferation in the context of bone marrow dysplasia and ineffective hematopoiesis.1 The members of this World Health Organization (WHO) designation include chronic myelomonocytic leukemia (CMML), atypical chronic myelomoncytic leukemia (aCML), juvenile myelomonocytic leukemia (JMML), and MDS/MPN unclassifiable (MDS/MPN-U).2 Refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T) remains a provisional entity under consideration for formal entry into the MDS/MPN designation by the WHO.
The underlying pathogenesis responsible for, or at least tolerant of, the bone marrow paradox is not clear nor is the molecular convergence point that biologically defines the MDS/MPN category as a whole. What is known is that the bone marrow phenotype of MDS/MPNs consists of increased cellular programmed cell death, resultant dysplasia, and cytopenias along with concomitant myeloid subset skewing and proliferation.3 This phenomenon is seen in MDS and MPNs, respectively, but not in combination. The annotation of recurrent genetic mutations in MDS/MPNs has led to significant insights into the dysregulated pathways that could be responsible for the combined phenotype (Table 1).4-7 As could be expected, mutations in signaling genes are recurrently mutated in MDS/MPNs such as CBL, JAK2, N- and K-RAS, and CSF3R in the background of “MDS-like” mutations, such as those of the spliceosome complex.4,7,8 These observations suggests that co-mutation of genes involved in dysplasia and bone marrow failure along with those of cytokine receptor signaling may, in part, explain the dual MDS/MPN phenotype. One example of comutation driving a MDS/MPN phenotype is that of RARS-T. A genetic hallmark of this MDS/MPN subtype is the comutation of SF3B1, a genetic determinant of ringed sideroblasts, and JAK2.9 This mutational combination occurs in approximately 40%-50% of all RARS-T but does not occur with the refractory anemia and ringed sideroblast (RARS) subtype of MDS or essential thrombocytosis in which SF3B1 and JAK2 mutations are common in isolation, respectively.10 Further, anecdotal reports have demonstrated that acquisition of JAK2 mutation is associated with a change from RARS to that of a RARS-T phenotype. This collectively suggests that these driver mutations are critical to the unique combinatory phenotype of RARS-T.
Frequencies of recurrent genetic mutations in MDS/MPNs
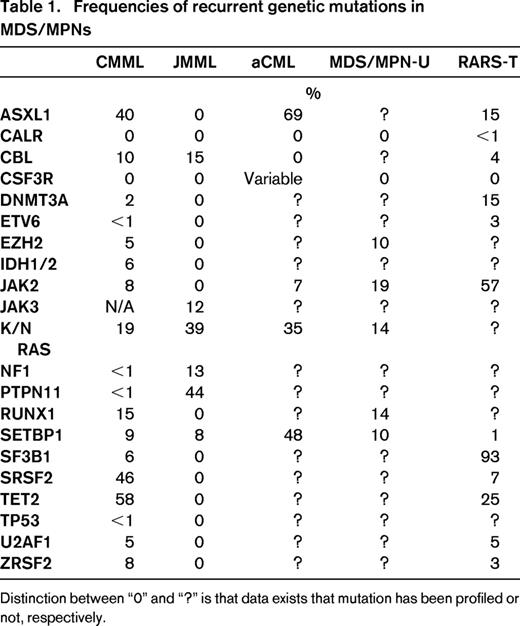
Distinction between “0” and “?” is that data exists that mutation has been profiled or not, respectively.
However, this mutational combination is not exclusively seen in MDS/MPN and not every MDS/MPN patient harbors a mutation in signaling related genes. Further, combinations of mutations that are not intuitively associated with myeloproliferation, such as that of SRSF2 and TET2 are highly associated with CMML.11 JMML, a monocytic leukemia seen in children <3 years of age, is associated with mutations that exclusively alter the RAS pathway without recurrent mutations in epigenetic or spliceosome genes.4 This suggests that, although some genetic combinations seen in MDS/MPNs are likely to explain bone marrow behavior, genetic alterations are unlikely to explain the major convergence point of MDS/MPNs in isolation. Deeper insights into the downstream consequences secondary to mutational combinations and their interaction with the bone marrow microenvironment will be critical to understanding the unique behavior of this class of malignancies.
Despite the phenotypic similarities in bone marrow behavior among MDS/MPNs, profound differences in the population affected, natural history, and clinical course exist. JMML is an uncommon myeloid malignancy of early life (age <3 yrs) with a disproportionate male preponderance. It is a heterogeneous clinical entity in that some patients, particularly those with Noonan syndrome, have spontaneous resolution of their disease despite identification of clonal hematopoiesis, whereas others can have a fulminant course.12 The natural history of CMML is also highly heterogeneous across patient subgroups with a median overall survival (OS) of 34 months and leukemic transformation rates of 15%-20% likely secondary to its genetic heterogeneity compared to JMML.13 Unlike JMML, CMML is a disease of the elderly with a median age of approximately 70 years and spontaneous remissions have not been reported. Atypical CML is a rare MDS/MPN whose incidence is estimated at 1% of that of typical, BCR-ABL1–positive CML.14 The natural history is not well established in the literature but the overall prognosis is thought to be poor. RARS-T is a comparably more indolent disease of the elderly with median survivals estimated as high as 7 years.10
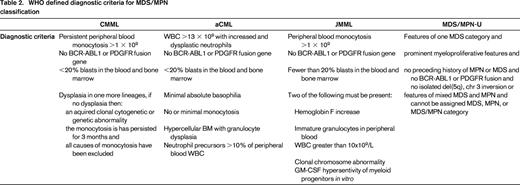
The WHO diagnostic criteria of MDS/MPNs are summarized in Table 2. In general, the respective MDS/MPN diseases are identified by the type of myeloid subset, which predominates in the peripheral blood. For example CMML and JMML are characterized by a unique expansion of peripheral blood monocytes, whereas aCML is associated with highly dysplastic granulocyte predominance.1,12 RARS-T is hallmarked by thrombocytosis while MDS/MPN-U has no clear association with a specific myeloid subset but instead is identified by clinical and/or pathologic manifestations of myeloproliferation and bone marrow failure.15 In this review, we will highlight the emerging preclinical tools, clinical data, and therapeutic strategies that are anticipated to increase our understanding of these unique diseases and improve their clinical management.
Bedside to bench or bench to bedside: MDS/MPN preclinical models
Models derived from human MDS/MPN genetic discoveries
Large-scale human genome sequencing in MDS/MPNs has led to advances in the annotation of recurrent genetic mutations that are postulated to be sufficient to induce the phenotype in which they were discovered. This, in turn, has led to the development of several monogeneic murine models that recapitulate a bone marrow failure syndrome or the MDS/MPN phenotype. For example, TET2 somatic loss of function mutations occur at frequencies approaching 50% in CMML and have be modeled by deleting TET2 in several murine systems.16 Interestingly, despite the wide-spread occurrence of TET2 mutations across myeloid malignancy, TET2 (−/−) murine models strikingly phenocopy proliferative CMML with features that include splenomegaly, monocytosis, and dysplasia.17,18 This is particularly notable given recent data demonstrating that early clonal dominance of TET2 mutations in human samples may be a CMML-specific feature suggesting that TET2 dosage may be a determinant of an MDS/MPN phenotype.19 Another example of a monogeneic MDS/MPN model informed by human genetics is ASXL1. ASXL1 frame shift mutations occur at comparable frequency to TET2 mutations and are universally adversely prognostic.20 In contrast to TET2, murine models deleting this gene [ASXL1 (−/−)] result in a bone marrow failure syndrome akin to MDS with concomitant genome wide alterations in H3K27me3 and H3K4me3 marks.21 SRSF2 conditional knock-in murine models have also been reported to induce a bone marrow failure syndrome and not MDS/MPN despite an incidence of SRSF2 mutations approaching 45%.22
NRAS, SHP2, NF1, and CBL mutations, which predominantly occur in JMML and CMML, have been modeled by several investigators and demonstrate a myelomonocytic phenotype similar to the above diseases.23-26 Interestingly, these murine models display profound GM-CSF-specific hypersensitivity which is a hallmark feature of JMML and CMML. Lastly, CSF3R murine models, which have been demonstrated to be mutated in aCML and chronic neutrophilic leukemia, have been generated. These mice indeed phenocopy an aCML- like disease and importantly respond to treatment with JAK inhibitors, an observation that was identified in in vitro human studies.27 Collectively these models are sufficient to induce a bone marrow disease that, in some cases, mimics MDS/MPN but suggests that complex interactions of gene mutations and perhaps microenvironmental influences are likely to explain genotype/phenotype relationships. It is anticipated that these models and their combinations will serve as excellent preclinical tools for therapeutic testing.
Models derived from preclinical observation
In addition to mice generated from observations from human disease, MDS/MPN phenotypes have been seen in murine models that were unexpected based on human genetic profiling. One interesting example is knockout of XIST. XIST is a well-described long-noncoding RNA (lncRNAs) that is critical for X chromosome inactivation. However, deletion of this non-coding RNA was sufficient to induce a highly penetrant and aggressive MDS/MPN in female mice.28 Hematopoeitic stem cells (HSCs) from this model demonstrated aberrant maturation and were of hematopoietic origin suggesting a protective effect of XIST. Another interesting model was that of NOTCH deficiency. NOTCH signaling suppression in murine models resulted in myelomonocytic predominance and a CMML-like disease. Further, mutations in NOTCH were identified, although rarely, in patient CMML samples suggesting human disease relevance.29 Other MDS/MPN models have been generated that were a result of CREBBP depletion, NR4A depletion, and others which have increased our understanding with respect to pathways whose dysregulation can lead to the disease phenotype.30,31
Diagnostic challenges and updates in MDS/MPNs
MDS/MPN by definition includes phenotypic properties of both MDS and MPNs, respectively. Thus, a critical diagnostic decision lies in determining that your patient does not fall in either of the above categories and is instead an overlap syndrome. The criteria to make these decisions in many cases are not straightforward. For example, patients may meet criteria for MDS but have pathologic evidence of overlap with megakaryocytic proliferation and clustering while having no clinical or peripheral evidence of proliferation. Should this patient be classified as MDS/MPN? Additionally, a patient may meet criteria for CMML but have subtle dysplasia and significant fibrosis in the bone marrow by reticulin staining.32 Should this patient be classified as myelofibrosis? Although diagnostic dilemmas occur at the interphase of diseases across cancer, it is particularly relevant to reflect on this in MDS/MPNs because its definition relegates the entire designation at the interphase between MDS and MPN. To this end, further research is needed to find MDS/MPN defining characteristics that can be objectively quantified and measured. It is tempting to hypothesize that recurrent gene mutations or, more likely, combinations of mutations may serve this purpose but further study is need. This is especially true in aCML and MDS/MPN-U because whereas monocytosis allows for the recognition of CMML and JMML, the differentiation between aCML and MDS/MPN-U from MPNs is more difficult.33
Disease-specific advances in the diagnostication of CMML have been reported. One key challenge for the diagnosis of CMML is discerning clonal from reactive monocytosis. This is important because the WHO diagnostic criterion of CMML does not require dysplasia, but can be diagnosed based solely on a persistent monocytosis that is unlikely to be caused by a concomitant condition. Because cytogenetic abnormalities occur in only 30% of cases, diagnosis is often delayed to ensure that monocytosis is persistent and expensive broad spectrum testing is often done to rule out concomitant conditions.13 However, recent data has demonstrated that sequencing only nine genes in the bone marrow or peripheral blood of CMML is sufficient to identify a clonal mark in 93% of cases.6 Given the increasing availability of targeted next generation sequencing myeloid panels that include these genes, our practice is to perform this test in the context of any suspected CMML case with diagnostic uncertainty.
Another key challenge is to differentiate CMML from other myeloid neoplasms with monocytosis. To this end, investigators have identified a monocyte subset restriction in CMML not seen in reactive monocytosis, other myeloid neoplasms with monocytosis, or aged-matched controls. This “repartitioning” of monocytes was validated using an external validation cohort and performed with high sensitivity and specificity for the diagnosis of CMML.34 Given that identifying this monocyte skewing is based on flow cytometry, translating this test to the clinical should be feasible and potentially incorporated in the diagnosis of this disease.
Prognostication updates for adult MDS/MPNs
Outside of RARS-T, the general prognosis of MDS/MPNs is poor. The median survival of CMML is reported to be 34 months and that of aCML is reported to be approximately 1 year.13 Although it is likely that significant heterogeneity exists among all adult MDS/MPNs, their rarity (aCML) and diagnostic uncertainty (MDS/MPN-U) has minimized efforts for risk stratification in some MDS/MPNs. However, significant efforts have been recently made in CMML and RARS-T.
The prognostication of CMML has been extensively investigated. At least 7 clinical models and 2 additional genetic models have been generated that incorporate ASXL1 mutation for the prognosis of CMML.35-41 Although these efforts have provided a rich resource for risk stratification, the number of models available has led to a lack of consensus among clinicians and researchers for the definition of “lower” and “higher” risk CMML. Further, the validity and relative prognostic power of these models had never been investigated in a large cohort precluding the rationale identification of a consensus model. To address this, we generated an international CMML database comprising >1800 cases for the purpose of identifying a consensus risk stratification model in CMML. Using this resource, we performed a detailed statistical analysis that was able to validate all models but were unable to identify a consensus model that was statistically superior. In fact, even after generating a novel model, we were unable to improve on the modest prognostic power of existing models. We next moved to interrogating the prognostic significance of gene mutation information and were able to confirm the independent prognostic significance of ASXL1 and identified CBL as independently prognostic in CMML.13 Efforts are now underway to add these gene mutations to existing clinical models so that a consensus prognostic score can be devised for CMML.
Although clinical anecdotes and small case series suggested that the natural history of RARS-T is relatively indolent relative to other MDS/MPNs, emerging data suggests that annotation of mutations in RARS-T may reveal a more aggressive subtype. A recent report identified SF3B1 and JAK2 mutations as favorably prognostic and associated with improved survival compared with their mutated counterpart. For example, SF3B1 wild-type cases of RARS-T had a median survival of 3.3 years compared to 6.9 years in mutated cases.10
Therapies in adult MDS/MPNs
There are no pharmacologic agents that have been demonstrated to alter the natural history of MDS/MPNs. As such, the general approach for the treatment of these diseases is to focus on therapeutic strategies tailored to individual patient symptomatologies. There is little evidence to suggest that asymptomatic MDS/MPN patients benefit from current standard treatment but it is strongly recommended that all MDS/MPN cases be considered for clinical trial when available. Allogeneic stem cell transplant has the most robust data suggesting a disease modify effect in MDS/MPNs and should therefore be considered in higher-risk MDS/MPN cases.42-45 Although there has been significant progress in the prognostication of CMML cases, challenges remain in adequately risk stratifying other MDS/MPNs for clinical transplant decision making.35,40,46 Below is a summary of treatment options and there disease specific supporting data.
Hypomethylating agents (HMAs) and noncytoreductive therapy
Robust data exists demonstrating the activity of HMAs in CMML. Several phase II studies have demonstrated that azacitadine is active in CMML and associated with acceptable therapy-associated toxicity.47 Decitabine has also been examined in multiple phase II trials, with response rates ranging from 10%-58% in patients with CMML.48 Advances have also been made that allow for improved prediction of response to decitabine in an individual CMML patient. A methylation signature was recently identified that can predict CMML overall response with high accuracy. This signature incorporated differentially methylated regions at nonpromoter sites and was validated in an external cohort.49-51 However, despite well documented activity, there exists no evidence that HMAs increase overall survival or decrease progression to AML. It is therefore our practice to reserve this therapy for CMML cases for which cytopenias are the predominant symptom. Very limited data is available for the use of HMAs for the treatment of aCML, MDS/MPN-U, or RARS-T. There is also limited data testing the activity of lenalidomide in RARS-T. Three case reports have demonstrated significant activity with respect to hematologic and splenomegaly improvement irrespective of cytogenetic abnormalities.51-53 Interferon has been tested in aCML in limited number of cases with modest activity.54,55
Cytoreductive agents
MDS/MPN cases with rapid and severe myeloproliferative or constitutional symptoms should be considered for treatment with cytoreductive agents. Several studies have reported the efficacy of the topoisomerase inhibitors topotecan and etoposide both as single-agent and in combination with cytarabine.56-58 Other trials have examined combination with arsenic or all-trans retinoic acid (ATRA) with modest results.59,60 The only randomized study that has been performed in CMML randomized 105 patients with CMML to receive hydroxyurea or etoposide which favored the hydrea arm.58 Induction chemotherapy has been used in CMML based on extrapolation from MDS. In our practice, we recommend induction chemotherapy in those cases with extreme or symptomatic leukocytosis, massive splenomegaly, or severe constitutional symptoms that are refractory to hydrea or other less intensive approaches.
Conclusions and opportunities
The MDS/MPNs are a unique class of myeloid malignancies that are coupled by a paradox in the bone marrow resulting in myeloid subset proliferation and bone marrow failure. Although the exact molecular convergence point is unknown, significant advances have been made secondary to a growing international community of MDS/MPN research. Although not fully annotated, the known spectrum of recurrent mutations in MDS/MONs have led to murine models that phenocopy MDS/MPNs and serve as useful tools for the molecular dissection of these diseases. Further, this genetic information has begun to augment our diagnosis and prognosis. It is anticipated that this information will additionally inform future therapeutic targets.
Although there are therapeutic options that have the potential to improve the symptomatology of MDS/MPN patients, the current landscape of pharmacologics is limited. Ongoing studies testing JAK2 inhibitors in aCML and CMML hold promise61 (NCT02092324 and NCT01776723), but only a small number of studies are currently addressing MDS/MPN cases specifically. Other strategies that may benefit the therapeutic armamentarium in MDS/MPNs are mutation-specific. For instance, trials using PI3K and MEK inhibitors are being tested in RAS mutated myeloid malignancies to include CMML. Taken together, MDS/MPNs are aggressive cancers that could, by any definition, be considered an area of unmet need in cancer research. Although they converge on a unique bone marrow phenotype, significant nonoverlaping characteristics exist that make it challenging to apply observations broadly across disease members. It is anticipated that disease specific study will be required to fully elucidate the underlying pathology of these malignancies but that scientific momentum, growing disease awareness, and advancing genomic tools will result in dramatic improvement in MDS/MPN clinical care.
Correspondence
Eric Padron, Department of Malignant Hematology, H. Lee Moffitt Cancer Center, 12902 Magnolia Dr, Tampa, FL 33612; Phone: 813-745-8212; Fax: 813-745-1720; e-mail: eric.padron@moffitt.org.
