
Abstract
Myelofibrosis (MF) is complex at the pathobiologic level and heterogeneous at the clinical level. The advances in molecular characterization of MF provide important insight into the mechanisms driving this chronic myeloid malignancy, refine risk stratification, offer novel therapeutic targets, and serve to measure therapeutic response. Although JAK2 inhibition has been the focus of laboratory and clinical efforts over the last decade, current experimental therapeutic approaches have broadened to include inhibitors of key alternative signaling pathways, epigenetic modulators, anti-fibrotics, and immunotherapies. Based on compelling preclinical rationale, a number of JAK2 inhibitor based combination therapies are now actively being evaluated in the clinic with the goal of disease course modification. The role and timing of hematopoietic stem cell transplant (HSCT) for MF has been challenged with the availability of commercial ruxolitinib and the plethora of experimental treatment options that exist. Integration of preconditioning JAK2 inhibition, reduced intensity conditioning regimens, and alternative donor sources are all being explored in an attempt to optimize this potentially curative modality. This review will summarize modern MF risk stratification, current clinical research approaches to chronic and advance phase MF focusing on novel agents alone and in combination, and update the reader on new directions in HSCT.
Learning Objectives
To understand the evolution of prognostication in myelofibrosis
To identify novel therapeutic approaches for the treatment of myelofibrosis
Myelofibrosis (MF) is a term that encompasses primary myelofibrosis (PMF), post-essential thrombocythemia (ET) and post-polycythemia (PV) MF (PET-MF, PPV-MF), and is considered the most severe subtype of the classic BCR-ABL1–negative myeloproliferative neoplasms (MPNs). MF is recognized by the World Health Organization (WHO) as a clonal hematopoietic stem cell (HSC) disorder characterized by myeloproliferation within the bone marrow, extramedullary hematopoiesis (EMH) leading to progressive organomegaly, anemia, and a leukoerythroblastic peripheral blood picture. MF is associated with a propensity to evolve into acute leukemia (predominantly of myeloid lineage) in 10%-20% of cases in the first decade after diagnosis. Generally speaking, chronic phase (CP) can be characterized by either a peripheral blood or bone marrow blast count <10%, accelerated phase (AP) of 10%-19%, and blast phase (BP) ≥20%. The prognosis for patients with MPN-BP is dismal (estimated at 3-5 months) and is tragically not improved with the use of induction regimens commonly used for acute myeloid leukemia (AML), unless followed immediately by hematopoietic stem cell transplantation (HSCT).1
It is also increasingly appreciated that MF is an intensely inflammatory state mediated by hyperactive JAK-STAT signaling (importantly, irrespective of JAK2 mutation status), which has been linked to the considerable symptom burden that results in reduced performance status and poor quality-of-life (QOL). The discovery of a point activating mutation in the JAK2 gene in 2005, renewed interest in both the academic and pharmaceutical communities in the study of MPNs and rapidly resulted in the first and only Food and Drug Administration (FDA)-approved therapeutic for MF, and more recently PV. However, realization of the importance of JAK2V617F in diagnosis, prognosis, and treatment of MPNs was tempered by the reality that much remained unknown of the molecular pathophysiology governing these myeloid malignancies. Subsequent studies have identified other driver mutations (MPL515L/K, CALR exon 9) associated with hyperactive JAK-STAT signaling, as well as numerous additional molecular events that influence signal transduction (CBL,LNK), chromatin modification (TET2, EZH2, IDH1/2, ASXL1), and RNA splicing (SF3B1, SRSF2, U2AF1), tumor suppressor function (TP53) and collectively contribute to disease progression and evolution. Moreover, recent investigations suggest that order of mutational acquisition can further influence MF phenotype and even response to ruxolitinib therapy in vitro.2 The current scientific appreciation of the complex clonal architecture of MPNs has provided the clinical basis for the current and future evaluation of single and combinations of agents targeting signaling pathways outside of the JAK-STAT pathway and epigenetic alterations. It is essential that current MF clinical trials incorporate mutational profiling at baseline to assess whether a mutational signature can predict therapeutic response.
Prior to discussing modern therapeutic advances in MF, it is worth updating the reader on the advances in risk stratification of MF patients in order to assess benefit/risk determination for experimental therapies and/or HSCT. The successes and deficiencies of current JAK2 inhibitor therapy for the treatment of MF must also be adequately reviewed before discussing ongoing trials evaluating agents directed at novel targets. Subsequently, this report will focus on next generation trials evaluating the combination of active agents from different therapeutic classes. Lastly, it is important to also acknowledge advances being made in HSCT as the only proven therapy to offer the possibility of cure in MF.
Refining risk stratification in MF
The first widely used risk stratification tool for MF was the Lille Classification which incorporated the following variables at time of diagnosis: white blood cell count (WBC) >30 × 109/L or <4 × 109/L earning 1 point and a hemoglobin <10 g/dL earning 1 point.3 Low risk (0 points), intermediate risk (1 point), and high risk (2 points) had associated median overall survivals of 93, 26, and 13 months, respectively. This risk stratification tool was simple and could be used universally as it only required a complete blood count. However, it was also clear that in so much as it was easy to utilize, it did not incorporate other important prognostic disease features that could better distinguish between groups of patients with differing outcomes. In 2009, Cervantes et al, on behalf of the International Working Group for MF Research and Treatment (IWG-MRT), set out to develop a PMF prognostication tool at time of diagnosis that would better discriminate prognosis and therefore provide more confidence in therapeutic decision making.4 The International Prognostic Scoring System (IPSS) incorporated five clinical features at the time of diagnosis that were found to have prognostic significance in multivariate analysis; age >65 years, presence of constitutional symptoms, hemoglobin <10g/dL, leukocytes >25 × 109/L, peripheral blood blasts ≥1%. The IPSS risk groups of low (0 points), intermediate-1 (1 point), intermediate-2 (2 points), and high (3+ points) provided nonoverlapping survival curves with median OS of 135, 95, 48, and 27 months, respectively. Shortly after the advent of the IPSS, Passamonti et al proposed the dynamic IPSS (DIPSS) which allowed application of a risk stratification system at any point during PMF disease course.5 The DIPSS risk groups used the same clinical variables as the IPSS, but more weight was assigned to anemia due to the higher prognostic power in time-dependent analysis. The DIPSS groups included low (0 points), intermediate-1 (1-2 points), intermediate-2 (3-4 points), and high risk (5-6 points) with a median OS not yet reached, 170, 48, 18 months, respectively. An age adjusted (aa)DIPSS was also created for PMF patients <65 years of age that would be traditionally considered appropriate for definitive therapy with HSCT. The DIPSS was later modified by the IWG-MRT to also include three additional negative prognostic risk factors including platelet count <100 × 109/L, the need for red blood cell transfusions, and the presence of an unfavorable karyotype (complex karyotype or sole or two abnormalities that include +8, −7/7q−, i(17q), −5/5q−, 12p−, inv(3), or 11q23 rearrangement).6 The DIPSS-plus stratifies patients into 4 risk groups with median OS of 185, 78, 35, and 16 months, respectively. Additionally, it was shown that an unfavorable karyotype and thrombocytopenia predicted leukemia-free survival (LFS) in this analysis with a 10 year risk of developing MPN-BP of 31% versus 12% in the absence of either risk factor.
Most recently, efforts to integrate cytogenomic information with the clinical prognostication systems have led to the development of the Genetics-Based Prognostic Scoring System (GPSS) and Mutation-Enhanced International Prognostic Scoring System (MIPSS; Figure 1.7,8 The mutational status of the three key drivers (JAK2, MPL, CALR) in addition to the presence of other somatic mutations (ASXL1, SRSF2, EZH2, and IDH1/2) that have shown to influence outcome were included in the construction of these newer prognostication systems.9,10 Age >60 years, hemoglobin <10g/dL, constitutional symptoms, platelets <200 × 109/L, “triple-negativity”, JAK2/MPL mutant, ASXL1 mutant, and SRSF2 mutant were all determined to have prognostic significance in multivariate analysis and used to develop 4 risk categories in a learning cohort. Low, intermediate-1, intermediate-2, and high-risk patients by MIPSS had OS of 26.4, 9.7, 6.4, and 1.9 years, respectively. When compared (using the Akaike information criterion) to the IPSS, the MIPSS provided refinement of the prognostic score and allowed the identification of subgroups of patients with a worse prognosis within an IPSS category. The genetics-based prognostic scoring system (GPSS) was developed by the Mayo group in a large cohort of PMF patients and validated in an independent patient cohort from Italy. The GPSS incorporates only cytogenetic and mutational prognostic data to create 4 risk groups of low, intermediate-1, intermediate-2, and high risk with corresponding OS of >17, 9, 5, and 2.2 years, respectively.

WBC indicates white blood cell; RBC, red blood cell; IPSS, International Prognostic Score System; DIPSS, dynamic IPSS; DIPSS+, DIPSS plus; MIPSS, Molecular International Prognostic Score System; and GPS, Genetics-Based Prognostic Scoring System. 1WBC <4 × 109/L or >30 × 109/L in Lille; WBC >25 × 109/L in IPSS, DIPSS, DIPSS+. 2Platelets <100 × 109/L in DIPSS+ and <200 × 109/L in MIPSS. 3DIPSS unfavorable karyotype: complex karyotype or 1 or 2 abnormalities including +8, −7/7q−, i(17q), −5/5q−, 12p−, inv(3) or 11q23 rearrangement; GPSS high-risk: complex without MK, 2 abnormalities not included in very high-risk category, 5q−, +8, other autosomal trisomies except +9, and other sole abnormalities not included in other risk categories; very high-risk: MK, inv(3), i(17q), −7/7q−, 11q or 12p. 4Includes driver mutation status (JAK2/MPL/CALR), as well as ASXL1 and SRSF2.
WBC indicates white blood cell; RBC, red blood cell; IPSS, International Prognostic Score System; DIPSS, dynamic IPSS; DIPSS+, DIPSS plus; MIPSS, Molecular International Prognostic Score System; and GPS, Genetics-Based Prognostic Scoring System. 1WBC <4 × 109/L or >30 × 109/L in Lille; WBC >25 × 109/L in IPSS, DIPSS, DIPSS+. 2Platelets <100 × 109/L in DIPSS+ and <200 × 109/L in MIPSS. 3DIPSS unfavorable karyotype: complex karyotype or 1 or 2 abnormalities including +8, −7/7q−, i(17q), −5/5q−, 12p−, inv(3) or 11q23 rearrangement; GPSS high-risk: complex without MK, 2 abnormalities not included in very high-risk category, 5q−, +8, other autosomal trisomies except +9, and other sole abnormalities not included in other risk categories; very high-risk: MK, inv(3), i(17q), −7/7q−, 11q or 12p. 4Includes driver mutation status (JAK2/MPL/CALR), as well as ASXL1 and SRSF2.
It is important to point out that the dizzying array of prognostication tools that have been created in the last few years have not been validated in PET-MF and PPV-MF and are still best utilized in the setting of determining eligibility for clinical trial enrolment and determining optimal benefit/risk ratio in the pursuit of HSCT. Presently, the DIPSS is most practical risk stratification tool available for the community practitioner in treatment decision making for their individual patient with MF.
A brief update on JAK2 inhibitors in MF
As a class of agents, JAK2 inhibitors have been very successful in reducing splenomegaly and ameliorating disease related symptoms. Patients treated with JAK2 inhibitors often feel better, attain an increase in physical mobility, and achieve a reversal of MF-related cachexia. Aspects of MF-associated biologic features such as potent reduction in levels of circulating inflammatory cytokines have been linked to the clinical improvement in constitutional symptoms following JAK2 inhibitor therapy.11 Although this may be a form of disease modification, the absence of molecular responses, and the lack of reproducible and convincing resolution of bone marrow fibrosis and MPN histopathologic features suggest that the malignant HSC clone itself is not sufficiently impacted by JAK-STAT inhibition alone.
The preclinical evaluation of the novel type II JAK2 inhibitor NVP-CHZ868 (Novartis), has been shown to inhibit JAK2 in the inactive conformation thereby inhibiting JAK-STAT signaling in JAK2 persistent MPN cells that have previously seen a type I JAK2 inhibitor.12 The proposed mechanism for this biologic phenomenon is discussed briefly in the combination therapy section below. NVP-CHZ868 appears highly active in an MPN murine model and may offer more potent inhibition of JAK-STAT signaling than currently tested type I inhibitors and deserves further evaluation in the clinic.
The 3 oral, small molecule, type I (competitive inhibitor of the adenosine triphosphate-binding site), JAK2 inhibitors with clear clinical activity and sufficient safety data to warrant discussion in 2015 are ruxolitinib (INCB18424, Incyte), pacritinib (SB1518, CTI biopharma), and momelotinib (CYT387, Gilead). A number of other JAK2 inhibitors (Table 1) have also been evaluated in clinical trials conducted in MF patients, some of which are no longer in development due to issues surrounding neurotoxicity (Fedratinib, Sanofi Aventis; XL019, Exelixis; AZD1480, AstraZeneca) and gastrointestinal toxicity (Lestaurtinib, Cephalon).13-15
JAK2 inhibitors tested in clinical trials in patients with myelofibrosis
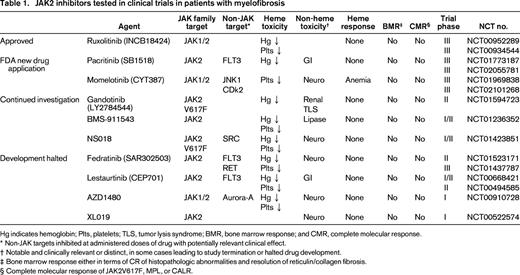
Hg indicates hemoglobin; Plts, platelets; TLS, tumor lysis syndrome; BMR, bone marrow response; and CMR, complete molecular response.
* Non-JAK targets inhibited at administered doses of drug with potentially relevant clinical effect.
† Notable and clinically relevant or distinct, in some cases leading to study termination or halted drug development.
‡ Bone marrow response either in terms of CR of histopathologic abnormalities and resolution of reticulin/collagen fibrosis.
§ Complete molecular response of JAK2V617F, MPL, or CALR.
Pacritinib is a JAK2/FLT3 kinase inhibitor that has completed phase I/II trials in patients with intermediate/high-risk MF.16-18 In a multicenter, single-arm, open-label, phase II trial of pacritinib in 35 patients with MF conducted in the United States, all patients had intermediate-1 or worse disease by DIPSS and 43% and 20% had baseline platelets <100 and <50 × 109/L, respectively. Grade 1-2 diarrhea (69%) and nausea (49%) were the most common nonhematologic toxicities, and led to treatment discontinuation in a only a single patient. Spleen responses were seen in 31% of patients by imaging and 42% by exam, and 58% of treated patients achieved symptom response. Grade 3/4 anemia and thrombocytopenia were seen in 26% and 20%, respectively. Importantly, drug interruption/discontinuation due to thrombocytopenia occurred in only 3 subjects. PERSIST-1 (NCT01773187) and 2 (NCT02055781) are randomized phase III trials comparing pacritinib to a best available therapy (BAT) arm aiming to provide efficacy and safety results in MF patients with low platelets as this remains a serious unmet clinical need. The results of PERSIST-1 were recently presented by Mesa et al as a late-breaking abstract at ASCO 2015. In this pivotal, randomized (2:1 pacritinib, BAT) phase III trial of 327 intermediate/high-risk MF patients, 32% and 15% had platelet counts <100 and <50 × 109/L. After a median duration of 16 months of pacritinib therapy, grade 3/4 adverse events were mainly restricted to nausea, vomiting, and diarrhea in <5%, 1%, and 1%, respectively. Hematologic toxicity was not different between the 2 arms and 25% of RBC transfusion dependent patients at baseline became independent with pacritinib therapy. Importantly, response rates were not diminished in the thrombocytopenic patients, with spleen volume reduction of ≥35% obtained in 17% versus 0% and 23% versus 0% in the pacritinib arm versus BAT arm with baseline platelets <100 and <50 × 109/L, respectively. Symptom improvement was also statistically significantly superior in the pacritinib treatment arm.
Momelotinib is another potent JAK1/2 inhibitor that was evaluated in a phase I/II trial in which long-term follow-up is available.19,20 This drug was effective in achieving combined spleen and symptom responses in 39% of the treated subjects. With a median treatment duration of 507 days, most interestingly and strikingly, 68% of transfusion dependent patients at baseline achieved independence for a minimum of 12 weeks and 26% of patients with baseline hemoglobin <10g/dL and not receiving transfusional support achieved at least a 2 g/dL increase in hemoglobin level. The median duration of the anemia response had not yet been reached at the time of this report. The pivotal phase III randomized trial comparing momelotinib to ruxolitinib in JAK2 inhibitor naïve patients is ongoing (NCT01969838), as well the phase III randomized study with BAT comparator arm in MF patients with cytopenias and prior ruxolitinib therapy (NCT02101268).
Ruxolitinib (Jakafi, Incyte) has been approved in the United States for the treatment of patients with intermediate/high-risk MF since 2011 and 3-year follow-up data from the 2 pivotal phase III trials, COMFORT-1 and 2 is available.21,22 It is essential to realize this indication includes patients with PMF as well as PET/PPV-MF and as of December 2014 has also been expanded to include patients with PV based on the results of the RESPONSE trial.23 Additionally, the package insert was subsequently updated to allow for the treatment of MF patients with a baseline platelet count as low as 50 × 109/L based on convincing safety data.24 The use of ruxolitinib in the treatment of PV is beyond the scope of this paper and is addressed elsewhere.25
There are 4 facets of ruxolitinib treatment in MF which need to be addressed and these include: (1) long-term safety, (2) durability of spleen and symptom response, (3) histopathologic changes in the bone marrow, and (4) improvement in overall survival. In terms of overall safety to date, with 3-year follow-up from the COMFORT studies reported, the emergence of late onset hematologic toxicity does not appear to be a major concern. The clinical relevance of maintaining a maximally reduced spleen volume for the entire duration of ruxolitinib treatment is unclear, as most patients will continue to report benefit from spleen related symptoms even if not at their maximum spleen volume reduction. However, the loss of symptom response with time will likely be clinically meaningful and prompt a change in therapy to either a next generation JAK2 inhibitor or JAK2 inhibitor-based combination therapy. It has even been suggested that ruxolitinib-treated MF patients with loss of response (either primary or secondary resistance) have a particularly poor outcome with a median survival of only 6 months.26 Exploratory analysis suggests that ruxolitinib induced regression/stabilization of bone marrow fibrosis in a subset of treated patients after prolonged exposure (60 months) is associated with down-regulation of certain inflammatory cytokines and modulation of the bone marrow microenvironment, but importantly, the actual clinical implication and significance of this finding is not entirely clear.27
A question that often arises, and for which no evidence-based data exists, is the use of ruxolitinib in MF patients with low-risk disease. Although there is a substantial amount of data to support the use of this agent in patients with intermediate/high-risk disease and even some suggestion of risk-score downgrading by some groups,28,29 there is simply no data to support earlier use in the disease course. Only a limited number of patients in the world have been treated beyond 5 years and the potential for infectious complications, disease progression, and loss of response at later time points is unknown and is therefore a serious concern in the low-risk group with an expected median survival that can exceed 15 years in some cases.
Non-JAK2 inhibitor therapy in MF
Based on an expanding understanding of the complex biologic mechanisms of MF pathogenesis, several novel therapeutic targets have emerged and multiple agents have already demonstrated preclinical as well as clinical activity. Agents that target alternative signaling pathways, epigenetic modifications, immune deregulation, HSC cell cycling, and bone marrow fibrosis are in various phases of clinical development (Table 2). In the last few years, the number of agents has grown substantially and precludes detailing each in this paper, as this has been done expertly in previous years.30 Instead, it is worth updating the reader of those with the most mature and otherwise remarkable results.
Novel non-JAK2 inhibitors in clinical trials for patients with myelofibrosis

HDAC indicates histone deacetylase; DNMT, DNA methyltransferase; HSP90, heat shock protein 90; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositide 3-kinase; PTX-2, pentraxin-2; IMiD, immunomodulatory; TGF-β, transforming growth factor beta; PD-1, programmed cell death-1; Epha3, ephrin type-A receptor 3; and IAP, inhibitor of apoptosis protein.
The first-in-class telomerase inhibitor, Imetelstat (GRN163L, Janssen), is a covalently-lipidated 13-mer thiophosphoramidate oligonucleotide that competitively binds the RNA component of human telomerase reverse transcriptase (hTERT).31 Rationale for the use of imetelstat in MPNs stems from observations that: (1) telomerase activity is increased in clonal granulocytes of patients with ET/PV, (2) telomere length is shortened in MPN patients and is directly correlated to the allelic burden of JAK2V617F, (3) imetelstat selectively induces apoptosis of MF CD34+ cells, and (4) selectively reduces malignant megakaryocyte progenitor cells.32-35 The investigator-initiated, single institution, pilot study of infusional imetelstat given every 3 weeks or weekly ×3 followed by every 3 weeks in 33 patients with intermediate/high-risk MF was first reported by Tefferi et al in 2013 and then updated in 2014 (NCT01731951).36,37 Remarkably, although 18 subjects terminated treatment for reasons of suboptimal response or progression, 7 patients responded (CR=4; PR=3) at a median treatment duration of 11 months. These responses included resolution of BM fibrosis and histopathologic abnormalities, as well as molecular responses. The investigators proposed that responses may be linked to mutational status as those patients with JAK2V617F, or SF3B1/U2AF1, and lacking ASXL1 mutation were more likely to attain a response. The numbers of treated patients were too few to conclude a clear connection between mutational status and treatment response, but do reinforce the importance of mutational profiling at baseline and at time of clinical response as a necessary correlative component of future trials. Grade 3/4 myelosuppression was frequent and transaminitis was an initial cause for concern prompting a brief clinical hold and FDA review. A highly anticipated company sponsored, multicenter, phase II trial in patients with MF as second-line therapy after ruxolitinib treatment will begin this year.
Another innovative approach that has attracted considerable attention is the Simon 2 stage, phase II trial of PRM-151 (Promedior) in patients with at least grade 2 bone marrow fibrosis (BMF) by European consensus classification. PRM-151 is a recombinant human pentraxin-2 (PTX-2) analogue, an acute phase response protein (serum amyloid P) that acts at sites of tissue damage, promoting wound healing, reducing inflammation, and inducing monocyte differentiation away from a fibrocyte and toward regulatory macrophage, resulting in reduction in the deposition of pathologic reticulin fibrosis.38 Serum PTX-2 levels have been shown to be reduced in patients with MF (as well as fibrotic diseases of kidney, lungs, and liver) and are inversely proportional to grade of BMF. Twenty-seven intermediate/high-risk MF patients with measureable BMF at baseline were treated on 4 study arms (PRM-151 at 2 different schedules with and without concurrent ruxolitinib).39 A total of 11 treated patients had documented reduction of BMF by at least 1 grade from baseline as assessed by central, blinded, adjudicated review. This infusional agent was well tolerated without any related grade 3/4 adverse events reported. Modest reduction in symptoms (as measured by the TSS), spleen size, and improvements in hemoglobin and platelet levels were observed and appeared to be independent of coadministration of the JAK1/2 inhibitor. A borderline association (p = 0.09) between reduction in bone marrow fibrosis and platelet response was appreciated with computer-assisted image analysis (CIA) performed on serial bone marrow slides.40 The second stage of this trial will be recruiting MF patients to PRM-151 monotherapy that are either ruxolitinib intolerant/resistant or naïve due to thrombocytopenia and will be conducted at multiple sites across the US and Europe (NCT01981850).
Ephrin ligands and their cognate receptor tyrosine kinases are bi-directional signalling pathways integral to cell positioning and tissue organization during normal embryonic development and their expression is lost in adult tissue. The ephrin receptor, EphA3, is selectively expressed in a broad spectrum of hematologic malignancies. EphA3 has been shown to be expressed specifically by leukemic stem cells, tumor neovasculature, and stroma, thus providing a novel therapeutic target for the treatment of hematologic malignancies.40,41 KB004 (KaloBios) is a recombinant, non-fucosylated IgG1k monoclonal antibody targeting the extracellular domain of EphA3 receptor kinase resulting in receptor activation, rounding of the cell and induction of apoptosis. KB004 exerts anti-leukemic activity via 4 distinct mechanisms: (1) antibody-dependent cell-mediated cytotoxicity (ADCC), (2) direct apoptosis, (2) disruption of the tumor vasculature, and (4) as an anti-fibrotic through a mechanism of action that remains unclear at present. The first-in-man dose escalation phase I/II of the trial in a broad spectrum of hematologic malignancies has concluded and the results were recently presented.42 Fifty patients with advanced hematologic malignancies were treated and other than grade 1/2 infusion reactions that were managed with premedication and slowing of the infusion rate, the agent appears to be well tolerated. Three responses (AML, MDS/MPN, and MF) were reported with the MF patient achieving a reduction in blast count, spleen size, BMF, and improvement in transfusion independence. Correlation between Epha3 expression level in the bone marrow and response remains exploratory. The dose expansion phase in the MF cohort at 250 mg IV weekly is currently underway at multiple centers (NCT01211691).
Focus on combination therapy in MF
The need to improve upon the current success of JAK2 inhibition in MF has also led to the evaluation of combination therapy in MF. The use of combinations of agents from different therapeutic classes to achieve deeper clinical responses is routine in other hematologic malignancies and is now an active strategy being pursued in MF. Unlike chronic myelogenous leukemia (CML) in which BCR-ABL1 as the primary disease driver can be effectively targeted by a single agent, MF will likely require polytherapy to achieve similar success. Presently, agents with proven biologic activity alone are being combined with ruxolitinib in an attempt to more effectively target the MPN HSC and achieve durable disease control. The specific rationale for each partner agent in combination is based either on preclinical laboratory investigation or in some cases, as an antidote to JAK2 inhibitor therapy induced myelosuppression.
Combination therapies that are currently being evaluated in MF clinical trials and are based on scientific rationale include ruxolitinib and epigenetic modifiers [panobinostat (NCT01693601, NCT01433445), azacitidine (NCT01787487), decitabine (NCT02076191); Table 3]. Combination therapy of ruxolitinib with immunomodulatory agents (IMiDs), such as lenalidomide (NCT01375140) and pomalidomide (NCT01644110) or the synthetic androgen, danazol (NCT01732445) or erythropoiesis-stimulating agents (darbopoeitin) are examples of partner agents chosen to counterbalance the therapy-related myelosuppression that is expected with ruxolitinib treatment. The first known attempt to formally evaluate the safety and clinical activity of triple-agent therapy in MF is being tested in a phase I trial with ruxolitinib in combination with the pan-PIM kinase inhibitor, PIM447, and the CDK4/6 inhibitor LEE011 (NCT02370706). This section will highlight several ongoing combination trials with both preclinical rationale and available clinical data.
Double- and triple-combination therapy trials in chronic and advanced phases of myelofibrosis
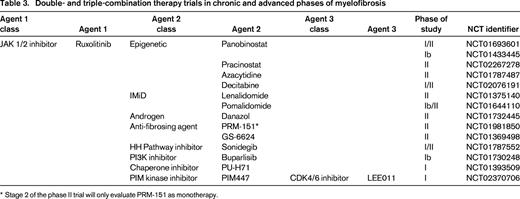
* Stage 2 of the phase II trial will only evaluate PRM-151 as monotherapy.
The combination of ruxolitinib and panobinostat is based on documented single-agent clinical activity in MF and synergistic activity in combination in a murine model of MF.43-45 The final results of the Novartis sponsored, multicenter, phase Ib trial conducted in Europe and involving 61 patients with intermediate/high-risk MF was reported at ASH 2014.46 The two oral drugs were very well tolerated in combination (diarrhea was the most common nonhematologic adverse event, grade 3/4 in 13%) and a maximal tolerated dose (MTD) was not achieved. The recommended phase II dose (RPTD) of ruxolitinib 15 mg twice daily continuously and concurrent oral panobinostat 25 mg thrice weekly every other week was established. At a median duration of ∼1 year exposure to the dual therapy at the RPTD, grade 3/4 anemia and thrombocytopenia was 32%, and 27%, respectively, and was the reason for discontinuation in 2 patients for each cytopenia. 85% of patients treated at the RPTD achieved a spleen reduction ≥50% and 59% attained nonpalpable splenomegaly. Reductions in JAK2V617F allele burden were modest, (29% of patients with mutation present at baseline had a ≥20% decrease from baseline). Three patients had reduction in BMF grade by blinded central review. An ongoing investigator initiated, single institution, phase I/II trial with a slightly different design of 1 month induction with ruxolitinib followed by combination therapy in patients with MF that can have had previous ruxolitinib exposure is currently enrolling in the US (NCT01433445).
Levine et al have postulated that chronic ruxolitinib exposure results in JAK2 transactivation allowing for continued JAK-STAT signaling. This mechanism for JAK2-dependent disease persistence in the presence of a JAK2 inhibitor provides rationale for additional or alternative targeting of JAK2.47 The concept that JAK2, an essential pharmacologic target in MF, is not effectively silenced with current type I JAK2 inhibitors has led to the preclinical evaluation of combination partner agents that alternatively target the protein. An example of this approach is combination therapy with inhibitors of heat shock protein 90 (HSP90) chaperone, in which JAK2 is a known client protein.48 JAK2 gene deletion studies in murine transplant models of MPL-driven MF have demonstrated significant reductions in clinical, bone marrow pathologic, and mutational burden when compared to JAK2 inhibitor therapy alone. Combined JAK1/2 inhibitor + the HSP90 inhibitor, PU-H71, in this murine model resulted in potent reduction of total JAK2 and phospho-JAK2 levels as opposed to increased phospo-JAK2 levels seen with JAK1/2 inhibitor therapy alone. Combination therapy led to more potent reduction in downstream JAK-STAT pathway mediator expression and provides the rationale for clinical evaluation of this combinatorial approach (NCT01393509). The single-agent HSP90 inhibitor, AUY922 (Novartis), is currently being evaluated in MF at a single institution, phase II trial in the US (NCT01668173), and future combination trials with this agent are also planned.
The Hedgehog (Hh) signaling pathway is active in various stages of normal hematopoiesis and has been shown to be up-regulated in primary MF granulocytes with significant increase in target gene expression of the transcription factor Gli1 and the tumor suppressor Ptch1.49 Additionally, the Hh-Gli1 pathway has also been implicated in the pathogenesis of fibrotic diseases and inhibition of this pathway is a therapeutic target in MF that was recently evaluated in a phase II trial of 14 patients with MF.50 Single-agent IPI-926 (saridegib; Infinity Pharmaceuticals), a smoothened (SMO) inhibitor was generally well tolerated without significant myelosuppression. However, only limited clinical responses and improvements in correlative biomarkers was observed and continued evaluation of this agent as monotherapy was terminated. In a murine transplant model of MF, the combination of ruxolitinib and the selective small molecule SMO inhibitor, sonidegib (LDE225, Novartis) resulted in synergistic activity in reducing leukocytosis, splenomegaly, BMF and mutant allele burden.49 A multicenter, phase Ib/II, combination trial of ruxolitinib and sonidegib in JAK2 inhibitor naïve patients with intermediate/high-risk MF is currently underway (NCT01787552).51 Results from the phase Ib portion of this trial have been presented and demonstrate tolerability and signal of clinical activity with grade 3/4 CPK elevation occurring in 10% of treated patients and 70% achieving ≥50% reduction palpable spleen.52 Sonidegib 400 mg daily in combination with ruxolitinib 20 mg twice daily is being explored for efficacy endpoints in the phase II portion currently.
Activity of phosphoinositide-3-kinase (PI3K)/AKT/ mammalian target of rapamycin (mTOR) pathway in MPN cells has been well documented and serves as a therapeutic target.53,54 Single-agent everolimus (RAD001, Afinitor, Novartis) was evaluated in a multicenter, phase I/I trial of 39 patients with MF and demonstrated a 23% response rate by IWG-MRT.55 Responses in symptomatology, spleen reduction and anemia were reported in the absence of changes in the JAK2V617F allele burden. Overall, everolimus was well tolerated and myelosuppression was modest and only reversible anemia was notable in this trial. Compelling preclinical studies utilizing the dual PI3K/AKT and mTOR inhibitor BEZ235 in combination with JAK2 inhibitors have demonstrated synergistic activity in a JAK2V617F-driven murine transplant model as well as against cultured and primary MPN CD34+ cells while sparing normal CD34+ hematopoietic cells.56,57 The pan-PI3K inhibitor buparlisib (BKM 120, Novartis) is being evaluated in combination with ruxolitinib in a multicenter phase Ib trial of patients with intermediate/high-risk MF (NCT01730248). Initial results were recently presented of this combination approach in 33 patients (ruxolitinib naïve and ruxolitinib resistant) in which the RPTD was determined to be ruxolitinib 15 mg twice daily and buparlisib 60 mg daily.58 The combination appears well tolerated with expected modest myelosuppression noted and reduction in spleen size in the majority of treated patients was observed. Perhaps most importantly, clinical activity was shown in patients who previously failed ruxolitinib monotherapy. Updated results with long-term follow-up are anticipated.
Role of HSCT in MF
HSCT remains the only proven therapeutic approach in MF that offers the potential for cure in those intermediate/high-risk individuals that are typically below 65-70 years of age, have a reasonable performance status, lack significant competing comorbid conditions, and importantly have an available related or unrelated HLA-matched donor. Considering these requisites, only a fraction of MF patients are appropriate candidates for HSCT. Efforts to address a high rate of non-relapse mortality (NRM) owing to the advanced age and high comorbid index of MF patients has been achieved through the use of reduced intensity conditioning (RIC) regimens. However, the success of reduction in transplant related mortality (TRM) associated with RIC preparative regimens can be compromised by an increase in risk of disease relapse.
A recent review from the Center for International Blood and Marrow Transplant Research (CIBMTR) of 233 MF patients who received RIC-HSCT revealed that the only independent predictor of poor outcome in multivariable analysis was the use of an unrelated donor source.59 This finding was also appreciated in the prospective, global, phase II MPD-RC 101 study of fludarabine/melphalan ± ATG-based RIC-HSCT in 66 MF patients. The rate of overall graft failure, acute graft versus host disease (aGVHD) grade II-IV, NRM, and 2-yr-OS was 6% versus 36%, 12% versus 21%, 22% versus 59%, and 75% versus 32% in the related versus unrelated arms, respectively.60 The exact reasons for such discrepant outcomes in these 2 arms is not readily apparent and was not due to differences in JAK2V617F status, age, degree of donor HLA match, or diagnosis (PMF or PET/PPV-MF). Unrelated donor source was also significantly associated with poor HSCT outcome due to a high rate of relapse and nonrelapse mortality in a large retrospective study conducted by the European Group for Blood and Marrow Transplantation.61
Given that 70% of patients do not have a matched related-donor option for HSCT, combined with the poor outcome reported in the unrelated arm of the MPD-RC 101 study, alternative donor sources need to be explored in MF, such as umbilical cord blood (UCB) and haploidentical (HI) donors. Although, prospective trials of HI-HSCT are currently lacking for patients with MF, a limited number of advanced phase MF patients have received this treatment at MD Anderson with encouraging results in terms of engraftment, graft versus host disease, and overall survival (Stefan Ciurea, MD Anderson, oral communication, June 11, 2015). The MPD-RC will be opening a phase II trial of RIC-HSCT using haploidentical donor grafts in transplant eligible MF and MPN-BP patients lacking HLA-matched related/unrelated donors (Table 4). A retrospective study of outcomes of UCBT for 35 MF/MPN-BP patients from European centers support the potential of this approach to improve survival in advanced phase MF, particularly in the subset of patients that received double-cord grafts in the setting of RIC regimen without the use of ATG.62
Current HSCT clinical trial approaches in myelofibrosis
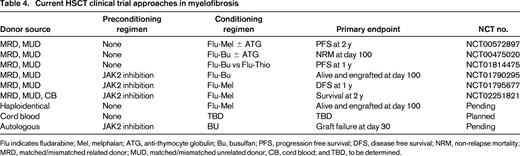
Flu indicates fludarabine; Mel, melphalan; ATG, anti-thymocyte globulin; Bu, busulfan; PFS, progression free survival; DFS, disease free survival; NRM, non-relapse mortality; MRD, matched/mismatched related donor; MUD, matched/mismatched unrelated donor; CB, cord blood; and TBD, to be determined.
Preconditioning therapy with a JAK1/2 inhibitor would be expected to improve performance status which may in turn reduce TRM, reduce splenomegaly which may improve donor cell engraftment, and suppress inflammatory cytokines which may modify GVHD.63 Based on this hypothesis, three prospective HSCT clinical trials are currently evaluating ruxolitinib therapy immediately in advance of conditioning for RIC-HSCT (NCT01790295, NCT02251821, NCT01795677) (Figure 2). The French Goelams-FIM/Sfgmtc group reported a preliminary description of their ongoing phase II JAK ALLO trial in which, alarmingly, cardiogenic shock (n = 2) and tumor lysis syndrome (TLS) complicated by acute renal failure (n = 1) occurred within 21 days of stopping ruxolitinib.64 The protocol was subsequently amended to allow for JAK1/2 inhibitor taper with prednisone overlay immediately prior to conditioning regimen and the use of TLS prophylaxis. Despite these measures, additional reports of 2 more cases of TLS (no renal failure) and 1 case of cardiogenic shock occurred within 2 weeks of ruxolitinib discontinuation. Conversely, retrospective analyses by others have not raised issues related to ruxolitinib withdrawal in the setting of HSCT.65,66 The MPD-RC 114 phase II RIC-HSCT trial is currently evaluating the safety and efficacy of at least 2 months of preconditioning JAK1/2 inhibition prior to fludarabine-based RIC regimen in intermediate/high-risk MF patients with related/unrelated donor options (Figure 2).

Rationale for JAK2 inhibition prior to HSCT in myelofibrosis. Splenomegaly contributing to hypersplenism-mediated sequestration of donor graft leading to primary graft failure, poor performance status and cachexia contributing to increased TRM, and hyper-inflammatory state contributing to increased risk of GVHD have all been identified as potential obstacles to successful outcome in HSCT of patients with MF. The use of a JAK2 inhibitor immediately prior to conditioning chemotherapy should result in reduction of splenomegaly, improvement in performance status, reversal of cachexia, suppression of circulating inflammatory cytokines. Therefore, the effects of JAK2 inhibition prior to HSCT is hypothesized to result in a decrease in rate of graft failure, TRM, and possibly GVHD.
Rationale for JAK2 inhibition prior to HSCT in myelofibrosis. Splenomegaly contributing to hypersplenism-mediated sequestration of donor graft leading to primary graft failure, poor performance status and cachexia contributing to increased TRM, and hyper-inflammatory state contributing to increased risk of GVHD have all been identified as potential obstacles to successful outcome in HSCT of patients with MF. The use of a JAK2 inhibitor immediately prior to conditioning chemotherapy should result in reduction of splenomegaly, improvement in performance status, reversal of cachexia, suppression of circulating inflammatory cytokines. Therefore, the effects of JAK2 inhibition prior to HSCT is hypothesized to result in a decrease in rate of graft failure, TRM, and possibly GVHD.
In the era of JAK2 inhibitors, the precise timing of HSCT is now unclear as the sense of urgency (patient and physician) can be delayed when responding to JAK2 inhibitor therapy and may result in a paradigm shift of delayed referral for evaluation of transplant only after progressive disease on a JAK2 inhibitor. Should HSCT be offered only upon loss of response to ruxolitinib, worsening treatment related cytopenias, or signs of progressive disease are important questions that still need to be answered.
Evaluating approaches to accelerated/blast phase MPN
MF in accelerated/blast phase (AP/BP) appears to be molecularly distinct from de novo AM, clinically more aggressive, and less responsive to traditional induction-remission chemotherapy.1,67 Although, an intensive therapy approach of induction chemotherapy followed by HSCT offers a reasonable potential of success for those that are appropriate candidates, alternative strategies that are less intensive, yet effective, are strongly desired.68 Due to the recognized role of hyperactive JAK-STAT signaling in MPN pathogenesis, a phase II trial of ruxolitinib monotherapy at doses of 25-50 mg twice daily was conducted in 38 AML patients in which 18 were MPN-BP and 3 CR/CRi were achieved in this group.69 More recently, higher doses of 50, 100, and 200 mg twice daily were tested in 26 patients with relapsed/refractory AML and a single CRi was reported with an increased rate of infectious complications observed.70 DNA methytransferase inhibitors (decitabine and azacytidine) have demonstrated clinical activity in retrospective series of MPN-BP.68,71,72 Using a JAK2V617F+/TP53 knock-out murine transplant model of MPN leukemic transformation, Rampal et al showed that ruxolitinib and decitabine inhibit colony formation in a dose-dependent fashion that was maximally enhanced in combination.67 This preclinical data in combination with previous documented clinical activity of decitabine has led to the MPD-RC 109 phase I/II trial of combination decitabine at the fixed dose of 20 mg/m2 × 5 days in combination with continuous ruxolitinib therapy in 28 day cycles, escalated in dose cohorts from 10 mg twice daily to 50 mg twice daily in patients with MPN-AP/BP (NCT02076191).
Conclusion
In the last 10 years since the discovery of JAK2V617F, significant advances have been made in terms of improved understanding and characterization of the molecular underpinning of the MPNs. It is clear that MF is a clinically heterogeneous disease process that is driven in most cases by at least three genetic alterations (JAK2V617F, CALR, MPL) and is further diversified by the acquisition of other genomic and epigenomic aberrations (ASXL1, TET2, EZH2, IDH1/2, SRSF2). The complex molecular pathogenesis of MF has thus far prohibited the successful use of a single targeted-agent approach to achieve molecular and histopathologic remissions, which are presumed to underlie achievement of improved PFS and OS. Rational therapies selectively targeting the malignant HSC and sparing normal hematopoiesis are currently being developed and evaluated in early phase clinical trials with the prospect of inducing remissions or “functional cures.” The evolution of MF risk-stratification tools integrating cytogenomic data will likely refine therapeutic decision making as available treatment options increase and particularly in calculating the benefit–risk ratio of HSCT (Figure 3). The next generation of combination therapy protocols in MF, novel treatment approaches to MF and MPN-BP, and development of more widely applicable HSCT options will undoubtedly serve to benefit our patients.

Modern risk-adapted treatment algorithm for myelofibrosis. The use of the dynamic international prognostic scoring system (DIPSS) and DIPSS-plus (DIPSS+) is the most comprehensive risk stratification tool currently available to guide therapeutic decision-making. The influence of driver mutation status as well the acquisition of additional somatic mutations may also prove to further refine this process. Ultimately, therapeutic approach is based on patient specific (age, performance status) and disease specific (symptom burden, presence of cytopenias) considerations in conjunction with an individual's personal goal of therapy. It should be stressed that whenever possible, clinical trial should be considered and hematopoietic stem cell transplantation remains the only potential curative treatment option available to a subset of patients. A indicates interferon-α; B, not FDA-approved for low-risk patients; C, second-generation JAK2 inh, combination therapy, imetelstat, PRM151, KB004, PF-04449913; D, HSCT on clinical trial when available; HU, hydroxyurea; RUX, ruxolitinib; and IMID, immunomodulator. *Erythropoiesis-stimulating agent can also be considered if baseline erythropoietin level is low.
Modern risk-adapted treatment algorithm for myelofibrosis. The use of the dynamic international prognostic scoring system (DIPSS) and DIPSS-plus (DIPSS+) is the most comprehensive risk stratification tool currently available to guide therapeutic decision-making. The influence of driver mutation status as well the acquisition of additional somatic mutations may also prove to further refine this process. Ultimately, therapeutic approach is based on patient specific (age, performance status) and disease specific (symptom burden, presence of cytopenias) considerations in conjunction with an individual's personal goal of therapy. It should be stressed that whenever possible, clinical trial should be considered and hematopoietic stem cell transplantation remains the only potential curative treatment option available to a subset of patients. A indicates interferon-α; B, not FDA-approved for low-risk patients; C, second-generation JAK2 inh, combination therapy, imetelstat, PRM151, KB004, PF-04449913; D, HSCT on clinical trial when available; HU, hydroxyurea; RUX, ruxolitinib; and IMID, immunomodulator. *Erythropoiesis-stimulating agent can also be considered if baseline erythropoietin level is low.
Acknowledgments
The author thanks Dr Ronald Hoffman for his enduring mentorship and critical review of the paper.
Correspondence
John Mascarenhas, Myeloproliferative Disorders Program, Tisch Cancer Institute, Division of Hematology/Oncology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Pl, Box 1079, New York, NY 10029; Phone: 212-241-3417; Fax: 212-876-5276; e-mail: john.mascarenhas@mssm.edu.
