
Abstract
The WHO classification provides the best diagnostic approach to myelodysplastic syndromes (MDS). However, biologic and analytic limitations have emerged in the criteria currently adopted to establish the diagnosis and to classify MDS. The provisional category of idiopathic cytopenia of undetermined significance (ICUS) has been proposed to describe patients in whom MDS is possible but not proven. To formulate a diagnosis of ICUS, a thorough diagnostic work-up is required and repeated tests should be performed to reach a conclusive diagnosis. Recent studies provided consistent evidence of age-related hematopoietic clones (clonal hematopoiesis of indeterminate potential; CHIP), driven by mutations of genes that are recurrently mutated in myeloid neoplasms and associated with increase in the risk of hematologic cancer. A subset of mutated genes, mainly involved in epigenetic regulation, are likely initiating lesions driving the expansion of a premalignant clone. However, in a fraction of subjects the detected clone may be a small malignant clone expanding under the drive of the detected and additional undetected mutations. In addition, several experimental evidences suggest the potential relevance of an abnormal bone marrow environment in the selection and evolution of hematopoietic clones in MDS. The spreading of massively parallel sequencing techniques is offering translational opportunities in the clinical approach to myeloid neoplasms. Although several issues remain to be clarified, targeted gene sequencing may be of potential value in the dissection between clonal myelodysplasia, nonclonal cytopenia, and clonal hematopoiesis arising upon aging or in the context of acquired marrow failure.
Learning Objectives
The biologic and analytic limitations of the current diagnostic approach to myelodysplastic syndromes (MDS), as well as the definition criteria and the clinical approach to the provisional category of idiopathic cytopenia of undetermined significance (ICUS)
The prevalence and clinical implications of hematopoietic clones arising upon aging (clonal hematopoiesis of indeterminate potential; CHIP), as well as in the context of acquired bone marrow failure, and the risk of hematologic cancer associated with these conditions
The most frequent somatic mutations driving age-related clonal expansions, and the biologic mechanism involved in the transition from clonal hematopoiesis to myeloid neoplasm, including clonal selection and evolution
Myelodysplastic syndromes (MDS) are defined in the World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues as a group of clonal hematopoietic stem cell diseases characterized by cytopenia, dysplasia in one or more of the major myeloid cell lines, ineffective hematopoiesis, and increased risk of progression to acute myeloid leukemia (AML).1
The current classification approach adopted by the WHO is based on a combination of morphology, immunophenotype, and genetic features to define distinct clinicopathologic disease entities, independently from the underlying causes that are often unknown.1 According to this principle, the diagnosis of MDS lies on 2 hallmarks, the evidence of myelodysplasia and the proof of clonal hematopoiesis. Myelodysplasia is a term used in pathology for describing morphologic abnormalities in one or more of the major hematopoietic cell lines.1 These abnormalities reflect the underlying pathophysiology; however, several factors may affect the circuitry that links the genetic lesions to the clinical phenotype, thus reducing the accuracy of the diagnostic process. The demonstration of the clonal nature of myelodysplastic hematopoiesis is an essential component of the WHO definition of MDS. However, according to our current knowledge most of the chromosomal abnormalities or discrete gene lesions detected in MDS are not specific and therefore unable per se to sustain a diagnosis of MDS.
This article will examine the borderland of the current diagnostic definition of MDS, and will discuss the biological clues and the clinical approach to conditions not fulfilling the criteria for diagnosis of MDS, as well as the clinical and biological implications of hematopoietic clones arising upon aging or in acquired cytopenias.
Current concept of the pathophysiology of MDS: clonal expansion of myelodysplastic stem cell, ineffective hematopoiesis, and leukemic transformation
According to the current understanding of the biology of MDS, the occurrence of a somatic mutation in an immature hematopoietic stem cell provides survival and growth advantage, leading to expansion of a clone. Once the myelodysplastic clone has become fully dominant in the bone marrow, the disease may become clinically apparent as a consequence of abnormal differentiation/maturation of clonal hematopoietic progenitors and precursors. Then, the acquisition of subclonal driver mutations involves the expansion of subclones, eventually leading to progression into acute leukemia.2
Recent studies showed that normal hematopoietic stem/progenitor cells from healthy subjects accumulate random mutations upon aging. Most of the mutations are random passenger events, whereas in most cases only one or two cooperating mutations are required to generate the malignant founding clone.3 According to this working model, in MDS the somatic mutation responsible for gain-of-function at the stem cell level involves loss-of-function in hematopoietic precursors; alternatively, myelodysplasia may result from a combination of somatic mutation(s) that provide a proliferative advantage to the stem cell and mutation(s) responsible for the defective differentiation/maturation.
Recurrent chromosomal abnormalities are detected in approximately one-half of patients with MDS. However, with the only exception of isolated del(5q), these are likely secondary genetic events, deriving from the genome instability caused by the founding genetic mutation.2 In the last few years, recurrent somatic mutations have been identified in several genes, including those of RNA splicing (SF3B1, SRSF2, U2AF1, ZRSR2), DNA methylation (TET2, DNMT3A, IDH1/2), histone modification (ASXL1, EZH2), transcription regulation (RUNX1), DNA repair (TP53), signal transduction (CBL, NRAS, KRAS), and cohesin complex (STAG2). Only 4-6 genes are consistently mutated in 10% or more MDS patients, whereas there is a long tail of ∼40 genes that are mutated less frequently (Figure 1).4-6

Frequency of recurrent somatic mutations in MDS, clonal hematopoiesis of indeterminate potential (CHIP) and aplastic anemia (AA).
Frequency of recurrent somatic mutations in MDS, clonal hematopoiesis of indeterminate potential (CHIP) and aplastic anemia (AA).
Diagnostic and classification criteria of myeloid neoplasms with myelodysplasia
The diagnostic approach to MDS includes morphologic studies of peripheral blood and bone marrow aspirate smears to evaluate abnormalities; bone marrow biopsy to assess marrow cellularity, topography and fibrosis; and cytogenetics to identify nonrandom chromosomal abnormalities. Additional investigations are also recommended, including flow cytometry immunophenotyping and fluorescence in situ hybridization (FISH).7
According to the WHO classification, MDS are currently categorized according to the number of dysplastic lineages, the presence of ring sideroblasts, and the percentage of blasts in the bone marrow and the peripheral blood.1 The WHO classification provides the best diagnostic approach to myeloid neoplasms with myelodysplasia; however, biologic and analytic limitations have emerged in the adopted criteria since their first proposal in 2001. In fact, the thresholds to define as significant the percentage of dysplastic cells, as well as of ring sideroblasts, are admittedly arbitrary. In addition, diagnosis and classification of MDS are compromised by the scarce reproducibility of morphologic analysis of dysplasia and by the poor specificity of dysplastic changes.8,9 Much effort has been made to standardize morphologic parameters,10 and to define minimal diagnostic criteria.11,12 Nonetheless, morphologic abnormalities can be detected in patients with nonclonal cytopenia, and in some noncytopenic controls.8 In addition, various studies showed a low rate of inter-observer concordance for assessment of dysplasia.13
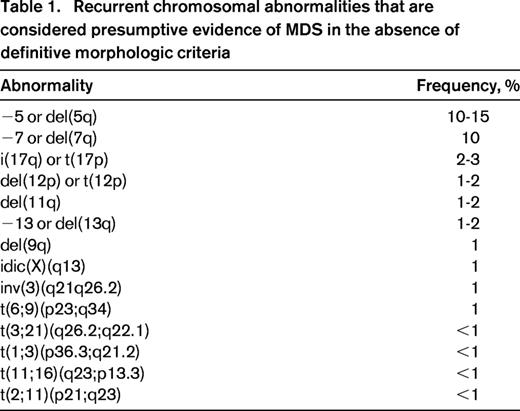
Recurrent chromosomal abnormalities are very important for the diagnosis of MDS, as they provide a marker of clonal proliferation. Some of the most frequent karyotypic aberrancies, such as +8, del(20q) and −Y, are not specifically associated with MDS. Conversely, according to the WHO 2008 criteria, selected abnormalities are recognized as presumptive evidence of MDS, even in the absence of definitive morphologic features (Table 1).1 Although these cases represent a minority of MDS, they provide the proof of concept that myelodysplasia may not be a required criterion for the diagnosis of MDS.
Clinical conditions at the boundary of MDS: idiopathic cytopenia of undetermined significance
Excluding or establishing the diagnosis of MDS may be challenging in the absence of robust morphologic criteria or a cytogenetic abnormality. In addition, an increasing number of patients with MDS are diagnosed at an early phase of disease, without prominent dysplasia at the bone marrow examination. To describe patients in whom MDS is possible but not proven, the term of idiopathic cytopenia of undetermined significance (ICUS) has been introduced.11 To be classified in this provisional category, patients must have relevant cytopenia in one or more lineage (hemoglobin <11 g/dL, neutrophil count <1.5 × 109/L, platelet count <100 × 109/L) that is persistent for at least 6 months, cannot be explained by any other disease and does not meet diagnostic criteria of MDS. To formulate a diagnosis of ICUS, a thorough diagnostic work-up is required to exclude hematologic and nonhematologic causes of cytopenia, and to inspect the diagnostic criteria for MDS. These patients should then be carefully monitored and repeated tests, including bone marrow examination, should be performed to reach a conclusive diagnosis.
Although the rationale underlying a provisional category of cytopenia of undetermined significance is unquestionable, the proposed definition has intrinsic limitations. In fact, whereas an adequate period of observation is required, even persistent cytopenia of shorter duration should be carefully considered if a diagnosis is not yet apparent. In addition, it must be acknowledged that, unlike other hematologic conditions of undetermined significance, such as monoclonal gammopathy of undetermined significance (MGUS) or monoclonal B cell lymphocytosis, a diagnosis of ICUS does not require the evidence of a clonal disorder.
Limited retrospective series of patients with ICUS were reported in the literature, providing conflicting results on the prevalence and outcome of this provisional category.14-16 These inconsistencies may be at least partly explained by the criteria applied to select the original study population and by high censoring rates due to incomplete follow-up. Larger prospective studies are required to estimate the prevalence of ICUS, to define their long-term outcome and their potential to progress to overt MDS or other hematologic neoplasm.
In the last few years, much effort has been made to develop new diagnostic tools able to increase the accuracy of the diagnostic approach to MDS, including flow cytometry immunophenotyping.17 According to the available studies, flow cytometry showed a sensitivity in identifying MDS ranging from 60% to 98%, with a specificity of 93% to 100%.18-21 In addition, in cases not fulfilling criteria for the diagnosis of MDS at the time of the first evaluation, flow cytometry showed a sensitivity up to 76% in patients receiving a final diagnosis of MDS, as well as a high negative predictive value.20,22 Although further studies are required to make diagnostic flow cytometry widely applicable in the diagnostic work-up of suspected MDS, the available evidence suggests that this approach has high sensitivity in cases with mild dysplasia by morphology.
Clinical implications of the genetic landscape of MDS
The identification of recurrent somatic mutations promised to have relevant implications for the diagnosis and classification of myeloid neoplasms with myelodysplasia, either by providing objective and reproducible markers of clonality and by identifying biologically homogeneous entities irrespective of morphologic diagnostic criteria.4,23
Within MDS, the only subtype currently defined by a genetic abnormality is MDS with isolated del(5q).1 Recently, a major step forward in genotype–phenotype correlation has been the identification of somatic mutations in SF3B1 in MDS with ring sideroblasts.24,25 SF3B1 mutation was proven to have a 98% positive predictive value for disease phenotype with ring sideroblasts, irrespective of the threshold of 15% for assignment to a sideroblastic subtype, suggesting that this threshold is not identifying separate entities.23,26 Although the identification of genetically-defined MDS entities is still developing, the available evidence clearly suggests that the recognition of disease entities based on specific genetic abnormalities has the potential to improve the accuracy of class prediction (ie, the process of determining which category a case belongs to), by providing robust biomarkers for diagnosis and classification regardless of morphologic criteria.
In addition, recent studies showed that approximately 90% of MDS patients carry one or more oncogenic mutations.4-6 Although most of the genes recurrently mutated in MDS can be detected in different myeloid neoplasms, they may be a valuable means of obtaining evidence of a clonal disorder in patients with suspected MDS.
Clonal hematopoiesis of indeterminate potential (CHIP)
The evidence of accumulating mutations in hematopoietic cells and of an increased risk of myeloid malignancies upon aging prompted investigations to screen healthy subjects for genes recurrently mutated in myeloid neoplasms using massive genome sequencing.
In the 1990s, landmark studies measuring X-chromosome inactivation pattern in females identified age-associated skewing in blood cells.27 More recently, a study by Busque et al identified TET2 mutations in a significant proportion of individuals with X-inactivation skewing without hematologic malignancy.28 This study provided the proof of concept that clonal hematopoiesis resulting from an expansion of cells harboring an initiating driver mutation is an aspect of the aging hematopoietic system, and suggested that these hematopoietic clones may represent a premalignant state.
More recently, three large studies provided consistent evidence of age-related hematopoietic clones, driven by mutations of genes that are recurrently mutated in myeloid neoplasms and associated with increase in the risk of hematologic cancer.29-31 A study from Xie et al analyzed blood-derived sequence data from 2728 patients with nonhematologic cancer from The Cancer Genome Atlas, and found recurrent blood-specific mutations in cancer-associated genes, the majority being associated with advanced age. Remarkably, most of these mutations were from recurrently mutated leukemia and/or lymphoma-associated genes, including DNMT3A, TET2, JAK2, ASXL1, TP53, and SF3B1 (Figure 1).29 Two subsequent studies analyzed data of peripheral blood cell DNA from 2 large cohorts of 12,380 and 17,182 persons, respectively, who were unselected for cancer or hematologic phenotypes.30,31 The results consistently showed an increase in the frequency of clonal somatic mutations with age. Mutations in genes implicated in hematologic cancers were found in up to 10% of persons 70-79 years of age, and 20% of persons 90 years of age or older (Figure 2).29-31

Prevalence of somatic mutations according to age and risk of development of a hematologic cancer among persons with somatic mutations. (A) Prevalence of somatic mutations according to age among persons not known to have hematologic disorders, estimated based on Xie et al,29 Jaiswal et al,31 Genovese et al,30 and McKerrell et al.32 Gray bands represent 95% confidence interval, calculated with binomial method. (B) Forest plot showing hazard ratios for development of a hematologic cancer among persons with somatic mutations who are not known to have hematologic disorders, according to Jaiswal et al,31 and Genovese et al.30 The upper part of the plot reports overall hazard ratio estimated from a meta-analysis of cohorts with the I2 statistic for the heterogeneity between studies, whereas in the lower part hazard ratios are reported according to the presence of candidate driver (CH-CD) or unknown driver (CH-UD) mutation, and to variant allele fraction (VAF of 0.10 or higher). Black diamonds indicate the HR for an individual cohort, while horizontal lines indicate the 95% confidence interval.
Prevalence of somatic mutations according to age and risk of development of a hematologic cancer among persons with somatic mutations. (A) Prevalence of somatic mutations according to age among persons not known to have hematologic disorders, estimated based on Xie et al,29 Jaiswal et al,31 Genovese et al,30 and McKerrell et al.32 Gray bands represent 95% confidence interval, calculated with binomial method. (B) Forest plot showing hazard ratios for development of a hematologic cancer among persons with somatic mutations who are not known to have hematologic disorders, according to Jaiswal et al,31 and Genovese et al.30 The upper part of the plot reports overall hazard ratio estimated from a meta-analysis of cohorts with the I2 statistic for the heterogeneity between studies, whereas in the lower part hazard ratios are reported according to the presence of candidate driver (CH-CD) or unknown driver (CH-UD) mutation, and to variant allele fraction (VAF of 0.10 or higher). Black diamonds indicate the HR for an individual cohort, while horizontal lines indicate the 95% confidence interval.
Detectable clonal expansions most frequently involved somatic mutations in 3 genes implicated in epigenetic regulation, DNMT3A, TET2, and ASXL1. In addition, other genes involved in myeloid neoplasms were consistently found to be mutated at a lower rate, including JAK2, SF3B1, SRSF2, and TP53 (Figure 1).29-31 Notably, a fraction of subject were shown to have clonal hematopoiesis without clear candidate driver mutations.30 The vast majority of subjects carrying detectable mutations had only one mutation in the set of examined genes (Figure 3), supporting the hypothesis that these persons had clones harboring only an initiating lesion. In addition, the median variant allele fraction for the identified mutations ranged from 9% to 19.5%, suggesting that these variants are present in only a subset of blood cells.29-31 A more recent study interrogated hot spot mutations at 15 gene loci recurrently mutated in myeloid malignancies using ultra-deep sequencing of blood DNA from blood donors or unselected individuals. As in previous studies, DNMT3A mutations were most common. Notably, mutations in splicing factors, SF3B1 and SRSF2, were identified only in subjects aged 70 year or older.32

Forest plot reporting number of somatic mutations (A) and allele burden (B) in MDS, AA, and clonal hematopoiesis (CH). Black diamonds indicate median values, whereas horizontal lines indicate the 95% confidence interval. For each disease the overall estimate resulted from a meta-analysis of cohorts (blue diamond) is also reported with the I2 statistic for the heterogeneity between studies. Data estimated from Papaemmanuil et al,4 Walter et al,5 Haferlach et al29 (MDS); Xie et al,29 , Jaiswal et al,31 Genovese et al,30 and McKerrell et al32 (CH); and Kulasekararaj et al36 and Yoshizato et al37 (AA).
Forest plot reporting number of somatic mutations (A) and allele burden (B) in MDS, AA, and clonal hematopoiesis (CH). Black diamonds indicate median values, whereas horizontal lines indicate the 95% confidence interval. For each disease the overall estimate resulted from a meta-analysis of cohorts (blue diamond) is also reported with the I2 statistic for the heterogeneity between studies. Data estimated from Papaemmanuil et al,4 Walter et al,5 Haferlach et al29 (MDS); Xie et al,29 , Jaiswal et al,31 Genovese et al,30 and McKerrell et al32 (CH); and Kulasekararaj et al36 and Yoshizato et al37 (AA).
Persons with clonal hematopoiesis had a risk of developing hematologic cancers elevated by a factor up to 13 than subjects without any detectable putative somatic mutations. This risk was further increased among persons with a variant allele fraction of 0.10 or greater (Figure 2).30,31 Overall, these figures translated into a risk of hematologic cancer of ∼0.5%-1% per year, which is not dissimilar to the risk of developing a plasma cell neoplasm in persons with MGUS. Carrying a mutation was also associated with increased all-cause mortality. Death from hematologic neoplasms alone could not account for the observed increase in mortality, whereas the reduced overall survival was associated with smoking and higher risk of death from cardiovascular causes.30,31
In the study by Jaiswal et al, no significant differences were noted in hemoglobin levels, white cell, and platelet counts in persons who had mutations in one or more genes compared with those who had no mutations. However, a significant increase in red-cell distribution width was observed in the subgroup of persons with mutations. In addition, subjects with multiple cytopenias were more likely to have mutations, and among persons with anemia, those with mutations had a higher percentage of unexplained anemia.31 Overall, these data suggest that most of the observed mutations do not influence significantly differentiation/maturation of hematopoietic cells, but that at least a subset of mutated genes or cooperating mutations may result in a perturbation of hematopoiesis. The term of clonal hematopoiesis of indeterminate potential (CHIP) has been introduced to define the condition characterized by the presence of a somatic mutation associated with hematologic malignancy in the absence of definitive diagnostic criteria for neoplasm.33
Aplastic anemia with clonal hematopoiesis
Acquired aplastic anemia (AA) is defined based on peripheral blood cytopenia and hypocellular bone marrow. The diagnosis of AA requires excluding other causes of pancytopenia; in particular, AA needs to be distinguished from hypoplastic MDS. This discrimination is compounded by the lack of clear-cut diagnostic criteria that may result in relevant diagnostic inconsistencies.34 In principle, cytogenetic and molecular genetic approaches may be a valuable criterion to distinguish hypoplastic MDS and AA. However, many observations provided evidence of clonal hematopoiesis in AA. In fact, ∼40% to 50% of patients with acquired AA have expanded clones of paroxysmal nocturnal hemoglobinuria (PNH) cells, as a result of somatic PIG-A gene mutations arising in hematopoietic stem cells. In addition, cytogenetic abnormalities, sometimes transient, have been reported in AA without MDS.35 The significance of clonal hematopoiesis in AA with regard to the development of a myeloid neoplasm, however, remain elusive.
Recently, targeted deep sequencing of genes implicated in myeloid malignancies was applied in patients with AA to investigate clonal hematopoietic populations.36,37 Kulasekararaj et al analyzed 150 acquired AA patients with no morphologic evidence of MDS, and detected somatic mutations in ∼20% of cases. Most frequently mutated genes included ASXL1, DNMT3A, and BCOR (Figure 1), with a median mutant allele burden of 20% (Figure 3). Patients with somatic mutations had shorter telomere lengths and higher risk of transformation to MDS compared with patients without mutations.36
More recently, targeted deep sequencing of candidate genes was performed in 439 patients with AA from a large collaborative study. Approximately one-third of patients had mutations in genes commonly affected in myeloid neoplasms. Most frequently mutated genes included PIGA, BCOR/BCORL1, DNMT3A, and ASXL1 (Figure 1). However, a substantial diversity was observed in mutation frequencies compared with myeloid neoplasms, mutations in PIGA, and BCOR/BCORL1 being more common in AA. In addition, mutations in AA showed significantly lower variant allele frequencies (<10% on average; Figure 3). Notably, whereas the size of DNMT3A or ASXL1 mutated clones tended to increase over time, that of BCOR/BCORL1 or PIGA mutated clones was more likely to decrease or remain stable. Furthermore, mutations in PIGA and BCOR/BCORL1 were associated with favorable survival, whereas other gene mutations were associated with worse survival and increased risk of progression to MDS or AML.37
Overall, these studies data provide evidence that a high proportion of AA patients with no morphologic evidence of MDS harbor mutations in genes typically seen in myeloid malignancies. Specific mutation patterns and clonal dynamics, however, strongly support the concept of selection of restricted cell clones under the pressure of an abnormal bone marrow environment.
Working models of transition from clonal hematopoiesis to myeloid neoplasm
Several lines of evidence suggest that CHIP or AA with clonal hematopoiesis and MDS may represent different steps of malignant transformation of hematopoietic clones. In fact, a subset of genes is consistently mutated across these conditions, suggesting that these mutations may represent the initial event of malignant transformation (Figure 1). In addition, the number of driver mutations per person and the mutant allele burden are increasing from CHIP and AA with clonal hematopoiesis to MDS (Figure 3), suggesting that these small hematopoietic clones either have the potential to progress to myeloid neoplasm or are small malignant clones at a preclinical stage. However, the mechanism by which each specific mutated gene is sustaining the transformation of the hematopoietic cell, as well as the role of factors external to the clone, such as bone marrow environment, are to be further clarified.
The available studies identified a subset of mutated genes, mainly involved in DNA methylation (DNMT3A, TET2) or chromatin modification (ASXL1), which are likely initiating lesions driving the expansion of a premalignant clone.28-31,36-38 The occurrence of additional cooperating mutations is then resulting in the transformation of the hematopoietic cell and in the expansion of a malignant clone (Figure 4A).

Models for the expansion of hematopoietic clones and the progression of into a hematologic cancer. (A) Expansion of a hematopoietic stem cell into a clonal population under the influence of a driver somatic mutation conferring a growth advantage to the cell, and potential progression into a myeloid neoplasm through the acquisition of subsequent subclonal driver mutations. (B) Expansion of a hematopoietic malignant clone under the influence of a driver mutation inferred from sequencing analysis and an undetected or as-yet-unknown cooperating mutation. In this scenario, the expected time to progression of the malignant clone to a clinical stage would be significantly shorter than under condition outlined in panel A. (C) Selection of a hematopoietic stem cell carrying a somatic mutation under the pressure of bone marrow environmental changes. The mutated clone is more fit to survive in an abnormal microenvironment than normal stem cells, and may expand sustaining a clonal hematopoiesis and eventually progress through the acquisition of subsequent subclonal driver mutations.
Models for the expansion of hematopoietic clones and the progression of into a hematologic cancer. (A) Expansion of a hematopoietic stem cell into a clonal population under the influence of a driver somatic mutation conferring a growth advantage to the cell, and potential progression into a myeloid neoplasm through the acquisition of subsequent subclonal driver mutations. (B) Expansion of a hematopoietic malignant clone under the influence of a driver mutation inferred from sequencing analysis and an undetected or as-yet-unknown cooperating mutation. In this scenario, the expected time to progression of the malignant clone to a clinical stage would be significantly shorter than under condition outlined in panel A. (C) Selection of a hematopoietic stem cell carrying a somatic mutation under the pressure of bone marrow environmental changes. The mutated clone is more fit to survive in an abnormal microenvironment than normal stem cells, and may expand sustaining a clonal hematopoiesis and eventually progress through the acquisition of subsequent subclonal driver mutations.
A fraction of subjects included in the studies on age-related clonal hematopoiesis had a very short timing from sampling to diagnosis of cancer,30 suggesting that the detected clone was a small malignant clone expanding under the drive of the detected and additional undetected or as-yet-unknown mutations, however, yet resulting in absent or mild abnormalities of blood counts at the time of analysis (Figure 4B).
Finally, the demonstration of distinct mutation patterns in AA provided evidence of the effect of Darwinian selection on survival and evolution of hematopoietic clones under the pressure of an abnormal bone marrow environment (Figure 4C).36,37 Several experimental evidences suggest that this mechanism could be relevant also in MDS. In fact, primary stromal dysfunction in mice was shown to promote MDS or AML.39 Recently, heterozygous conditional knock-in of Sf3b1K700E mutation resulted in a clinical phenotype consistent with MDS, but in competitive repopulation assays Sf3b1K700E hematopoietic stem cells did not show a proliferative advantage.40 Interestingly, in the study by McKerrell et al, adopting ultra-deep sequencing in unselected individuals, mutations in SF3B1 and SRSF2 were selectively identified in subjects aged 70 year or older, suggesting that selection pressure related to the aging of hematopoietic system may contribute to the expansions of clones harboring these gene mutations.32
Practical approach to unexplained cytopenia in the era of genomic medicine
The spreading of massively parallel sequencing techniques is offering translational opportunities in the clinical approach to myeloid neoplasms. Although several issues about the significance and the tumoral potential of clonal hematopoiesis remain to be clarified, the potential value of gene sequencing in the dissection between clonal myelodysplasia, nonclonal cytopenia, and clonal hematopoiesis arising upon aging or in the context of acquired marrow failure, is tangible.
Several studies reported a high prevalence of anemia in the elderly, and in about one-third of these conditions evident causes of anemia cannot be identified (unexplained anemia or cytopenia). At present, these subjects should be carefully monitored and repeated tests, including bone marrow examination, should be performed to reach a conclusive diagnosis. In case of unexplained anemia/cytopenia, a screening of recurrent somatic mutations on DNA from peripheral blood cells might be of potential value. In fact, the negative predictive value in case of absence of detectable mutations in the most common candidate driver genes appears to be sufficiently high to advocate avoiding or at least postponing further bone marrow investigation. It must be noted, however, that the available studies identified a fraction of MDS patients without known driver mutation,4-6 as well as a fraction of subjects with clonal hematopoiesis with unknown drivers,30 suggesting that subjects with undetected mutations may however deserve a careful clinical follow-up. Conversely, subjects with evidence of clonal cytopenia would be legitimately candidate to bone marrow investigations to inspect the diagnostic criteria for MDS. Confidently, future studies correlating mutation status and bone marrow morphologic findings in unselected subjects with unexplained cytopenia will allow identifying specific mutations or mutation patterns that may be considered as presumptive evidence of MDS, even in the absence of definitive morphologic features.
Although the available studies on clonal hematopoiesis showed that persons with detectable mutations have a risk of hematologic cancer of ∼1% per year,31 at present a screening of unselected individuals without hematologic phenotype seems premature.
It must be acknowledged, however, that prospective studies including detailed hematologic examinations and careful follow-up are warranted to prove the diagnostic value of a screening of somatic mutations in patients with suspected MDS. Not negligibly, although the costs of massively parallel sequencing technology are expected to progressively decrease in the next future, cost-effectiveness analyses should be also taken into consideration before validating such approaches.
Correspondence
Luca Malcovati, Department of Molecular Medicine, University of Pavia Medical School and Department of Hematology Oncology, Fondazione IRCCS Policlinico San Matteo, 27100 Pavia, Italy; Phone: +39-0382-503062; Fax: +39-0382-502250; e-mail: luca.malcovati@unipv.it.
