
Abstract
The anemia of chronic disease is an old disease concept, but contemporary research in the role of proinflammatory cytokines and iron biology has shed new light on the pathophysiology of the condition. Recent epidemiologic studies have connected the anemia of chronic disease with critical illness, obesity, aging, and kidney failure, as well as with the well-established associations of cancer, chronic infection, and autoimmune disease. Functional iron deficiency, mediated principally by the interaction of interleukin-6, the iron regulatory hormone hepcidin, and the iron exporter ferroportin, is a major contributor to the anemia of chronic disease. Although anemia is associated with adverse outcomes, experimental models suggest that iron sequestration is desirable in the setting of severe infection. Experimental therapeutic approaches targeting interleukin-6 or the ferroportin–hepcidin axis have shown efficacy in reversing anemia in either animal models or human patients, although these agents have not yet been approved for the treatment of the anemia of chronic disease.
Learning Objectives
To define the human diseases that associate with the anemia of chronic disease
To delineate the role of inflammatory cytokines and the iron regulatory hormone hepcidin in causing the anemia of chronic disease
To understand the potential role for novel therapies to treat the anemia of chronic disease
Epidemiology
The anemia of chronic disease refers to the impaired production of erythrocytes associated with chronic inflammatory states, including cancer, chronic infection, or autoimmune diseases. Recent data indicate that anemia can also occur in the setting of severe, acute inflammation, such as critical illness, or with milder but persistent inflammatory signals that occur in obesity, aging, and kidney failure. For these reasons, the name “anemia of inflammation” may be more suitable than anemia of chronic disease. The National Health and Nutrition Examination Study (NHANES III) revealed that ∼1 million Americans older than 65 years exhibit anemia related to inflammation. In NHANES III, anemia of inflammation was defined as a low serum iron level (<10.74 μM or <60 μg/dL) without evidence of low iron stores, ie, transferrin saturation >15%, serum ferritin >12 ng/mL, or erythrocyte protoporphyrin concentration >1.24 μM.1 Other features of anemia of inflammation include inappropriately low levels of erythropoietin and elevated measures of inflammatory markers, such as C-reactive protein.2
The anemia of inflammation is particularly common in hospitalized patients. In the CRIT study of anemia and blood transfusion in the critically ill, an observational cohort analysis of 4892 patients in intensive care units across the United States,3 mean hemoglobin levels in these patients decreased over a 30-day period despite the administration of blood transfusions. Furthermore, a nadir hemoglobin <9 g/dL was an independent predictor of increased mortality and length of stay. In a recent study of 191 consecutive hospitalized elderly patients with anemia, 70% of patients were found to have anemia of chronic disease. Sixteen percent of the patients with anemia of chronic disease had concomitant chronic renal failure.4 Seventy-one percent of the patients with anemia of chronic disease were suffering from an acute infection, 12% had cancer, and 16% had a chronic infection, such as a pressure ulcer, or a chronic autoimmune inflammatory disease.4
The intersection of obesity, chronic inflammation, and metabolic derangements with anemia is an emerging area of interest. Obese patients exhibit higher plasma levels of proinflammatory cytokines and acute-phase reactants, as well as higher rates of iron-restricted erythropoiesis that can result in anemia.5 In patients with chronic inflammation, one would expect increased levels of serum ferritin, thus the concept of “functional iron deficiency” has been defined for patients with serum ferritin <100 ng/mL despite chronic inflammation.6 A recent cross-sectional study of 947 obese patients under evaluation for bariatric surgery revealed that 52.5% exhibited functional iron deficiency, defined as a serum ferritin of 12-100 ng/mL for females or 15-100 ng/mL for males with serum C-reactive protein >3 mg/L.7 Seventy percent of obese patients with functional iron deficiency exhibited a transferrin saturation <20%.7 Further suggesting a link between obesity and impaired iron metabolism, weight loss has been associated with an increase in transferrin saturation in overweight individuals.8
Anemia commonly affects patients with neoplasia. Although hematologic cancers are more likely to cause anemia via infiltration of the bone marrow by an abnormal cell population, solid tumors often cause anemia even without bone marrow involvement. A recent prospective, observational study of 888 patients with a variety of carcinomas revealed that 63.4% of the patients were anemic.9 The prevalence and severity of anemia increased with a more advanced stage of cancer. Furthermore, advanced-stage patients had significantly increased mean plasma levels of markers of inflammation, including C-reactive protein, fibrinogen, interleukin (IL)-6, tumor necrosis factor α, IL-1β, ferritin, hepcidin, erythropoietin, and reactive oxygen species, compared with early-stage patients, whereas serum iron, leptin, triglyceride, and glutathione peroxidase levels were reduced significantly in advanced-stage patients.9
Pathophysiology
The discovery of the iron exporter ferroportin and the peptide hormone hepcidin revolutionized the understanding of anemia of inflammation. As erythroid progenitors mature to the polychromatophilic stage, they express increasing amounts of transferrin receptor 1 to acquire iron for the production of hemoglobin. Macrophages consume senescent erythrocytes, degrade hemoglobin, and store the liberated iron in ferritin for subsequent release of iron to developing erythrocytes.10 Characterization of the ferroportin-deficient zebrafish weissherbst11,12 and the ferroportin knockout mouse13 revealed that ferroportin is required for the export of iron from macrophages to developing erythrocytes. In ferroportin-deficiency, iron remains sequestered in macrophages, resulting in impaired delivery to developing erythrocytes and a block in erythroid maturation.11,12
In response to iron overload or inflammation, human hepatocytes secrete the iron-regulatory peptide hormone hepcidin.14-16 Hepcidin binds ferroportin, resulting in the internalization and degradation of both proteins17 (Figure 1). Hepcidin appears to be the key regulator of iron homeostasis, because loss-of-function mutations in genes that regulate Hepcidin expression, for example, Transferrin receptor 2, HFE, Hemojuvelin, or in Hepcidin itself, have each been associated with hereditary iron overload syndromes.18 Maintaining appropriate hepcidin levels depends on a complex interaction of regulatory factors, including bone morphogenic proteins (BMPs), the BMP coreceptor hemojuvelin, proteases such as furin and matriptase-2, and inflammatory cytokines.

Inflammation stimulates increased production of the iron-regulatory peptide hepcidin by hepatocytes. Hepcidin binds the iron exporter ferroportin (Fpn), causing internalization and degradation of both proteins and decreasing delivery of iron from macrophages to developing erythrocytes. This impairs erythroid development and leads to anemia.
Inflammation stimulates increased production of the iron-regulatory peptide hepcidin by hepatocytes. Hepcidin binds the iron exporter ferroportin (Fpn), causing internalization and degradation of both proteins and decreasing delivery of iron from macrophages to developing erythrocytes. This impairs erythroid development and leads to anemia.
Inflammatory cytokines that trigger Hepcidin expression include IL-6 and IL-1β,19 whereas transcription factors that mediate the effects of inflammation include Stat3, C/EBPα, and p5320,21 (Figure 2). IL-6 enhances JAK/Stat signaling,22,23 which leads to increased phosphorylation of Stat3 and increased Stat3 binding to the Hepcidin promoter.21,24 IL-1β induces Hepcidin expression via the C/EBPα and BMP/SMAD signaling pathways. Hepatocyte damage, via endoplasmic reticulum stress or oxidation, enhances C/EBPα25,26 or Stat3 activity,27 respectively, and increases Hepcidin expression. Lipopolysaccharide (LPS) released by severe bacterial infection activates toll-like receptor (TLR) 4 signaling, which enhances production of IL-628 by macrophages. IL-6, in turn, stimulates hepatocyte production of hepcidin.

Inflammation and hepatocyte damage augment Hepcidin transcription and iron sequestration via several pathways. LPS released by bacterial infection activates TLR-4 signaling, which increases IL-6 release by macrophages. IL-6 signaling leads to phosphorylation of Stat3 and increased Stat3 binding to the Hepcidin promoter, whereas endoplasmic reticulum (ER) stress in hepatocytes promotes CEBP-α binding to the Hepcidin promoter. BMP signaling via ligands, such as BMP2, BMP4, BMP6, and BMP9 activating BMP receptor-I, causes Smad phosphorylation and Smad binding to the Hepcidin promoter, which is required for Hepcidin transcription. The BMP coreceptor hemojuvelin (HJV) interacts with the BMP receptor to enhance BMP signaling. Inflammation also promotes macrophage release of lipocalin, which can interact with bacterial siderophores to sequester iron.
Inflammation and hepatocyte damage augment Hepcidin transcription and iron sequestration via several pathways. LPS released by bacterial infection activates TLR-4 signaling, which increases IL-6 release by macrophages. IL-6 signaling leads to phosphorylation of Stat3 and increased Stat3 binding to the Hepcidin promoter, whereas endoplasmic reticulum (ER) stress in hepatocytes promotes CEBP-α binding to the Hepcidin promoter. BMP signaling via ligands, such as BMP2, BMP4, BMP6, and BMP9 activating BMP receptor-I, causes Smad phosphorylation and Smad binding to the Hepcidin promoter, which is required for Hepcidin transcription. The BMP coreceptor hemojuvelin (HJV) interacts with the BMP receptor to enhance BMP signaling. Inflammation also promotes macrophage release of lipocalin, which can interact with bacterial siderophores to sequester iron.
Hepcidin is not the only protein causing iron sequestration during bacterial infection. Recent studies indicate that stimulation of TLR2 and TLR6 in mouse models reduces expression of ferroportin in macrophages and causes hypoferremia without increasing macrophage hepcidin expression.29 LPS stimulates macrophages to produce lipocalin 2, which sequesters iron by binding bacterially produced siderophores.30 Furthermore, infection or inflammation stimulates neutrophil release of the iron binding protein lactoferrin, which can be internalized by bacteria, isolate iron from pathogens, and arrest microbial growth.31
Obese individuals exhibit increased plasma levels of proinflammatory cytokines, the metabolic regulatory hormones leptin and hepcidin, and the iron-sequestering protein lipocalin-2. There are two proposed mechanisms by which obesity may contribute to functional iron deficiency and anemia based on experimental models: (1) leptin and proinflammatory cytokines stimulate hepcidin production in adipocytes and hepatocytes32 ; and (2) adipocytes and peripheral blood mononuclear cells in obese patients produce lipocalin 2, which restricts iron availability to developing erythroid cells.33 In addition to effects on iron metabolism, proinflammatory cytokines diminish erythropoietin synthesis, impair the differentiation of erythroid progenitors, and shorten the lifespan of mature red blood cells.34
New experimental models
New models of the anemia of chronic disease are facilitating the evaluation of therapeutic approaches to the condition. The killed Brucella abortus model of anemia of inflammation35,36 produces a 50% decrease in hemoglobin level in mice 14 days after a single intraperitoneal injection. Furthermore, erythropoiesis gradually recovers after the injection, just as human patients may recover from the insult of a critical illness, such as sepsis. The ease and consistency of the killed Brucella abortus model provided the basis for an in-depth evaluation of the hematologic effects of inflammation over time and in multiple mouse strains. Injection of the killed Brucella abortus bacteria in Hepcidin knockout mice produced a blunted effect on the onset of anemia. Gardenghi et al.35 further evaluated the effects in IL-6 knockout mice and found that IL-6 deficiency protected against hypoferremia and anemia. Arguing for a protective effect of the anemia of inflammation, Hepcidin knockout mice exhibited significantly impaired survival compared with wild-type controls after injection of killed Brucella abortus.36
Experimental treatments
The best treatment for the anemia of chronic disease is to eradicate the underlying disease. When that is not possible, patients have been treated with transfusions, intravenous iron supplementation, and erythropoiesis stimulating agents.37 Newer, experimental approaches target IL-6 activity and the hepcidin–ferroportin axis. The first observations indicating that IL-6 is a potential drug target for the anemia of chronic disease were documented in 1993, when researchers observed that treating patients with metastatic renal cell carcinoma with a murine anti-IL-6 antibody improved paraneoplastic thrombocytosis and anemia.38,39
Multicentric Castleman disease (MCD) has proven to be a useful human model of the anemia of chronic disease and a testing ground for therapies directed against IL-6. Patients with MCD exhibit generalized lymphadenopathy, systemic chronic inflammation, increased IL-6 activity, and anemia of inflammation.40 Siltuximab, a human–mouse chimeric anti-IL-6 antibody was developed and approved for the treatment of MCD but has also been shown to improve anemia in patients with this condition.41 Investigators hypothesized that siltuximab would be effective in early-stage myelodysplastic syndrome in which levels of proinflammatory cytokines are often elevated, but siltuximab failed to reduce transfusion dependence in a phase 2 trial in patients with transfusion-dependent, low- or intermediate-risk myelodysplastic syndrome.42 Tocilizumab, a humanized monoclonal antibody against the IL-6 receptor, has been shown to reduce serum hepcidin levels and improve anemia in patients with MCD and rheumatoid arthritis.37,40 Tocilizumab has now been approved to treat rheumatoid arthritis.
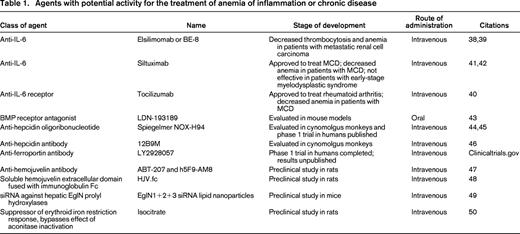
Efforts to modulate the ferroportin–hepcidin axis have included drugs that inhibit hepcidin production, agents that block the activity of the hepcidin peptide, and antibodies to ferroportin. LDN-193189 is a potent BMP receptor antagonist that has been shown to inhibit BMP signaling and reduce hepcidin mRNA levels in a dose-dependent manner when administered to mice by oral gavage. Furthermore, LDN-193189 ameliorated the anemia of inflammation in mice that had been injected with turpentine to generate sterile abscesses.43 This agent is of particular interest because it is an orally available small molecule. Other efforts have focused on parenterally administered agents. NOX-H94 is a structured l-oligoribonucleotide that binds human hepcidin with a high affinity. In cynomolgus monkey models, NOX-H94 blocked IL-6-induced hypoferremia and moderated the development of anemia.44 In human trials, NOX-H94 blocked LPS-induced hypoferremia.45 The human anti-hepcidin antibody 12B9M increased serum iron levels in cynomolgus monkeys but has not been evaluated in a primate model of anemia of inflammation.46 A phase 1 trial to evaluate the effect of an antibody against ferroportin, LY2928057, has been completed, but the results are not yet available (clinicaltrials.gov). Table 1 summarizes other experimental therapies in development.
Agents with potential activity for the treatment of anemia of inflammation or chronic disease
Our knowledge of the interaction between inflammation, iron metabolism, and erythropoiesis has improved our ability to understand the anemia of chronic disease. Although drugs have not yet been approved to treat this condition, several agents are under investigation, and some agents improve anemia of inflammation in patients with MCD or rheumatoid arthritis. In some cases, the anemia of inflammation may be protective; for instance, in animal models of the anemia of critical illness, hepcidin-deficient mice exhibited significantly lower rates of survival than wild-type animals.36 Thus, the best course of action continues to be to identify and treat the underlying causes of the anemia of chronic disease.
Correspondence
Dr Paula G. Fraenkel, Beth Israel Deaconess Medical Center, Division of Hematology/Oncology and Cancer Research Institute, 330 Brookline Avenue, CLS 434, Boston, MA 02215-5491; Phone: 617-667-2168; Fax: 866-345-0065. E-mail: pfraenke@bidmc.harvard.edu.
