
Abstract
Relapsed and refractory leukemias pose substantial challenges in both children and adults, with very little progress being made in more than a decade. Targeted immunotherapy using chimeric antigen receptor (CAR)-modified T cells has emerged as a potent therapy with an innovative mechanism. Dramatic clinical responses with complete remission rates as high as 90% have been reported using CAR-modified T cells directed against the B-cell-specific antigen CD19 in patients with relapsed/refractory acute lymphoblastic leukemia. Supraphysiologic T-cell proliferation, a hallmark of this therapy, contributes to both efficacy and the most notable toxicity, cytokine release syndrome, posing a unique challenge for toxicity management. Further studies are necessary to identify additional targets, standardize approaches to cytokine release syndrome management, and determine the durability of remissions.
Learning Objective
To describe factors leading to successful and highly active cell therapies and the risk factors for toxicity
Introduction
Relapsed acute lymphoblastic leukemia (ALL) is a leading cause of cancer deaths in children and has a dismal prognosis in adults.1-3 Chemotherapy intensification has largely been responsible for dramatic improvements in survival for pediatric ALL,4 but ∼1 in 5 children with ALL will relapse despite current intensive treatment regimens and most of these children will not survive.1,3 Nearly 85% of children in first relapse will achieve a second complete remission (CR2); however, those remissions are frequently not sustained.1,3 Salvage therapy for second or greater BM relapse induces remissions in only 40% of patients and long-term survival is quite poor.3 Overall survival for adults with ALL is poor (30%–40%),5 and induction of CR2 remains quite difficult in adults, with rates of 50% at best.2 Poor salvage rates suggest that many of these leukemias are refractory to conventional cytotoxic chemotherapy, necessitating novel approaches. In recent years, genomic characterization has guided the study of therapies targeting leukemogenic lesions,6,7 which were driven by the success of imatinib in Philadelphia chromosome (Ph)-positive ALL8 ; however, driver lesions can be found in only a subset of ALL.
Immune-mediated elimination of tumor cells has long been recognized and is the basis for both cancer vaccines and cellular therapies, including hematopoietic stem cell transplantation (HSCT). Adoptive transfer of T cells engineered to express a chimeric antigen receptor (CAR) is emerging as an extremely powerful technology for the treatment of chemotherapy-refractory leukemias.9-11 By combining the specificity of a monoclonal antibody with the activation domains of T cells, CARs deliver activated T cells with potent cytotoxicity to antigen-expressing tumor cells. This chapter reviews this mode of targeted immunotherapy for leukemia, with particular attention on CD19-directed CARs, the best studied and most advanced CAR T cell therapy to date.
Design and mechanism
CARs were first described >20 years ago as a means of introducing tumor specificity into adoptive cell therapy.12 The principle of antigen-specific T cell therapy was realized with first-generation CARs, which link an antibody-derived single-chain variable fragment (scFv) to the CD3ζ intracellular signaling domain of the TCR complex (Figure 1). Subsequent modifications incorporated 1 (second-generation CAR) or 2 (third-generation CAR) costimulatory domains, with the first costimulatory signal supplied by CD28 or 4-1BB and the second by other costimulatory molecules such as CD27, CD28, 4-1BB, ICOS, or OX40 (Figure 1).13-15

CAR structure. CAR molecules link an extracellular scFv to intracellular signaling domains. The intracellular component includes the CD3ζ intracellular signaling domain of the TCR either alone (first-generation) or in combination with 1 (second-generation) or 2 (third-generation) costimulatory domains. (Image courtesy of M. Maus.41 )
CAR structure. CAR molecules link an extracellular scFv to intracellular signaling domains. The intracellular component includes the CD3ζ intracellular signaling domain of the TCR either alone (first-generation) or in combination with 1 (second-generation) or 2 (third-generation) costimulatory domains. (Image courtesy of M. Maus.41 )
Autologous T cells are engineered to express CARs through various gene transfer technologies. Retroviral and lentiviral vectors allow for long-term expression through permanent genetic modification. The advantage of this approach is the potential for long-term disease control from a single infusion of engineered T cells. However, ongoing on-target toxicity and the theoretical risk of transformation are potential concerns.16,17 Transient expression can be achieved with RNA insertion through electroporation18 and may be desirable in some circumstances, for example, when on-target toxicity or integration into the genome is a concern. RNA-based approaches can produce substantial tumor responses; however, expression beyond ∼1 week requires repeated infusions,19 so long-term disease control may still be possible with this approach but would require multiple treatments.
Regardless of the method of gene transfer, in vitro cell culture systems for T cell expansion are used to manufacture large quantities of engineered T cells (Figure 2). These systems use antibodies and/or various artificial APCs to engage CD3 and activate T cells, with costimulation provided by a second signal or cytokine.20 Depending on the method, the manufactured product may contain different proportions of memory-type T cells, with the potential for significant replicative capacity, and potent effector T cells, which are terminally differentiated and have minimal replicative capacity.

Manufacture of CAR-modified T cells. Peripheral blood mononuclear cells collected by leukapheresis are expanded ex vivo and transduced to express the CAR molecule before infusion into the patient. In this example, magnetic beads coated with antibodies to CD3 and CD28 are used for ex vivo expansion.40 (Image used with permission from Novartis Pharmaceuticals. Copyright 2014, Novartis Corporation.)
Manufacture of CAR-modified T cells. Peripheral blood mononuclear cells collected by leukapheresis are expanded ex vivo and transduced to express the CAR molecule before infusion into the patient. In this example, magnetic beads coated with antibodies to CD3 and CD28 are used for ex vivo expansion.40 (Image used with permission from Novartis Pharmaceuticals. Copyright 2014, Novartis Corporation.)
Through its scFv domain, CAR-modified T cells recognize and bind the target antigen with antibody-like specificity, which allows for high-affinity recognition of cell-surface proteins. Once engaged, CARs link activated T cells to malignant cells expressing the target antigen, triggering a cell-mediated immune response that bypasses the MHC. Engagement can lead to a cytotoxic T-cell response as well as massive T-cell proliferation in vivo.
Leukemia targets
The ideal target for CAR-modified T cells would be universally expressed on tumor cells but not expressed on normal cells. Because such targets are rare, antigens that are minimally expressed on normal cells or expressed on cells with functions that are dispensable or replaceable represent viable targets. CD19 has emerged as an attractive target due to its specificity for one cell lineage (B cells) and near universal expression on B-cell malignancies, including chronic lymphocytic leukemia (CLL), ALL, and many non-Hodgkin lymphomas.21 Additional B-cell antigens, such as CD22, are under active investigation and show potential.22
T-cell leukemias pose a particular challenge for target identification because leukemic blasts express the same antigens as normal T cells. Although individual leukemias may aberrantly express antigens that are not normally expressed on T cells, there is no universal target that is specific to T lymphoblasts.
Target discovery for myeloid leukemias is also problematic because the antigen profile overlaps with hematopoietic stem cells.23 Although CARs directed against CD123 have demonstrated efficacy in preclinical models,24,25 expression of CD123 on vascular endothelial cells will require investigation prior to safe translation to the clinical setting. CD33 is a potential target, but anti-CD33 CAR T-cell therapy would require transient CAR expression or allogeneic stem cell rescue.
CD19 CAR clinical trials: outcomes and toxicity
Due to its limited expression profile, CD19-directed T-cell therapies for B cell leukemias have led the way for CAR clinical trials. Since the first demonstration of clinical efficacy in CLL,11 which was associated with exponential in vivo proliferation and long-term persistence, other groups have confirmed these results with distinct CAR designs.13,26 Our group and others have extended these findings, showing a very high degree of clinical activity in ALL.9,10,27 Clinical trials of CD19 CARs are being conducted at several institutions and were well-represented at the 55th ASH annual meeting in 2013 (Table 1).
CD19 CAR clinical trials presented at the 55th ASH annual meeting in 2013*
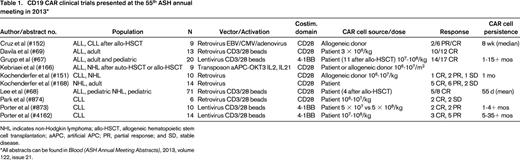
NHL indicates non-Hodgkin lymphoma; allo-HSCT, allogeneic hematopoietic stem cell transplantation; aAPC, artificial APC; PR, partial response; and SD, stable disease.
All abstracts can be found in Blood (ASH Annual Meeting Abstracts), 2013, volume 122, issue 21.
Initial reports of a small number of patients showed dramatic responses in refractory, bulky CLL, which then was extended to relapsed, highly refractory ALL.9-11 With larger studies and more mature follow-up, CAR-modified T cells were shown to persist for >3 years in CLL patients with an initial response rate of 57% and a CR rate of 29%,28 which compares favorably to ibrutinib (overall response rate of 71% but CR rate of 2.4%).29 More recently, studies have demonstrated extraordinary CR rates in ALL. Davila et al recently reported an 88% CR rate in a cohort of 16 adults with relapsed B-ALL.30 Similarly, we have reported a 90% CR rate in 30 patients (25 on a pediatric study and 5 on an adult study) with relapsed/refractory ALL treated in Children's Hospital of Philadelphia and University of Pennsylvania phase 1 trials.31
Long-term outcomes
Comparable initial responses have been demonstrated by several groups; however, long-term outcomes may be discrepant. Although CR rates are much higher in ALL, CAR T-cell persistence appears to be shorter in some patients with ALL compared with CLL patients who respond.30 This discrepancy may be related to the different kinetics of tumor growth and elimination of the 2 diseases, as well as the antigen reservoir provided by bulky disease characteristic of CLL. Davila et al reported 2-3 months of 19-28z CAR T-cell persistence in their ALL cohort, with ∼1/2 of patients proceeding to allogeneic stem cell transplantation (SCT).30 Long-term outcomes were not reported on those not proceeding to SCT. We have demonstrated longer persistence (up to 24 months) of CTL019 T cells, which use the 4-1BB costimulatory domain, in ALL patients.31 Continued B-cell aplasia, also seen out to 1-2 years in some patients, suggests continued effector function of CAR T cells. Interim analysis of 30 ALL patients treated with CTL019 demonstrates a 6-month event-free survival of 67% and overall survival of 78%. Furthermore, ongoing remissions for up to 2 years are possible in the absence of SCT. For many pediatric patients with refractory disease who enter CR1 or CR2, allogeneic SCT is the standard of care, although many of our patients in this situation have elected not to proceed to SCT. However, 2/3 of our patients already have undergone allogeneic SCT, relapsed, and then achieved remission with engineered T-cell therapy. In these patients, recommending a second (or later) SCT is open to discussion.
Cytokine release syndrome
Toxicities associated with CAR-modified T-cell therapies are related to the exponential T-cell proliferation that is necessary for efficacy, making toxicity management a balance of risks. Cytokine release syndrome (CRS) is the most common and prominent toxicity. CRS is an inflammatory process associated with considerable elevations in cytokine levels, as its name suggests.32 CRS symptoms range from mild flu-like symptoms, including high fevers and myalgias, to severe vascular leak and hypotension potentially resulting in multiorgan system failure. In our recent series, all of our patients experienced some degree of CRS, including those with low disease burdens. However, 27% experienced severe CRS, characterized by hypotension in all such patients and acute capillary leak and respiratory distress in many. Severe CRS occurred only in patients with higher disease burdens.31 Clinical symptomatology, laboratory abnormalities (including profound hyperferritinemia), and cytokine elevation pattern all mimic hemophagocytic lymphohistiocytosis,10 a rare disorder of immune regulation leading to pathologic cellular activation and inflammation.33 Although corticosteroids and etoposide are the mainstays of hemophagocytic lymphohistiocytosis therapy, both destroy T cells and would almost certainly affect clinical efficacy.
We have observed substantial IL-6 elevation during peak T-cell proliferation and demonstrated a rapid and profound reversal of life-threatening CRS, without apparent effect on CAR T-cell efficacy, after administration of tocilizumab, a monoclonal antibody to the IL-6 receptor.10,31,32 A similar pattern, including a rapid response to tocilizumab, was seen in CRS caused by the bispecific antibody blinatumomab.35 After this initial report, we and others have now incorporated tocilizumab into the management of severe CRS, with rapid and dramatic responses.34,36
The only reproducible predictor of severe CRS identified currently is high disease burden at infusion.30,31 C-reactive protein (CRP) has been proposed as an indicator of severe CRS.30 Although severe CRS is associated with higher CRP levels, we have observed high CRP in patients without severe CRS, suggesting that this indicator may or may not apply to all CAR designs.31 Cytokine levels may be better indicators, but are not readily available in a clinically useful timeframe in most institutions.34 Further studies are needed to develop algorithms for identification of at-risk patients and perhaps prevention of life-threatening CRS with earlier treatment.
Encephalopathy
Neurologic toxicity has been reported with CAR-modified T-cell therapies, as well as another T-cell-engaging therapy, blinatumomab.30,37,38 The primary neurologic toxicity is global encephalopathy, but seizures have been reported as well.30 The pathophysiology of encephalopathy is unknown, but may be inflammatory in nature and may be related to high cytokine levels. However, because we have seen encephalopathy appear immediately after resolution of CRS and after administration of tocilizumab, it may not be preventable by IL-6R blockade. In our experience, encephalopathy is self-limited, with resolution over several days without intervention or apparent long-term sequelae.
B-cell aplasia
Although CD19 is close to an ideal target due to limited expression on normal tissue, it is expressed throughout B-cell development. Therefore, CD19-directed therapies eliminate the B-cell lineage, producing an off-tumor, on-target toxicity. With long-term persistence of T cells that express an anti-CD19 CAR, B-cell aplasia will almost certainly continue as long as the CARs continue to function. In our experience, this has extended past 2 years in children and 3 years in adults. In the recent cohort, the probability of engineered T cells persisting at 6 months after infusion, as measured by flow cytometry, was 68% and the probability of relapse-free B-cell aplasia was 73% at the same time point.31 Immunoglobulin replacement mitigates infectious complications; however, longer follow-up is needed to assess late toxicity of B-cell aplasia.
Challenges and future directions
For CAR T-cell therapies to reach their full potential, several challenges need to be addressed. The first is extension across 2 different arenas: diseases and institutions. For CAR T-cell therapies to be viable in other disease types, tumor-specific targets must be identified or modifications to treatment strategy or design made. Although target identification is an area of active investigation, promising targets remain elusive for many diseases.39 Transient expression is one option for antigens with undesirable tumor to normal cell profiles. Another option is combination or tandem CARs, which join 2 antigen-recognition moieties, allowing for specific recognition of antigens expressed together on tumor cells but not on normal cells.
For CAR T-cell therapies to be feasible across institutions, comprehensive training of clinicians and a standardized approach to CRS management will be necessary to maximize safety.34 A related challenge is minimizing severe toxicities. Across studies, disease burden appears to be a reliable predictor of CRS severity in responding patients. However, decreasing disease burden before CAR T-cell therapy often is not possible, but may become more feasible as CAR therapy is deployed earlier in the disease course. Laboratory predictors of CRS severity, such as cytokine levels or CRP, are under investigation and may lead to an algorithm for earlier intervention in at-risk patients. To be clinically actionable, cytokine levels will need to be widely available in real time. Nevertheless, stressing the importance of severe CRS prediction presumes a viable intervention exists. Tocilizumab has proven effective during maximal symptomatology. It remains to be determined if earlier tocilizumab administration can prevent life-threatening CRS and do so without compromising efficacy.
Finally, despite the impressive responses to CAR T-cell therapies to date, relapse remains a challenge. Although patients with CD19+ disease recurrence resulting from short CAR T-cell persistence may respond to retreatment with CAR cells, patients who relapse with CD19− disease and chemotherapy-refractory disease may not have further treatment options. Future work aimed at optimization of CAR design or T-cell-replicative properties may prevent some of these relapses by prolonging T-cell persistence. Escape variants leading to CD19− relapses are potential and real concerns of therapies with solo targets. We have seen several patients relapse with CD19− disease, including the second patient we treated in our phase 1 study.10 CARs directed against other antigens, such as CD22, may salvage some of these relapses and are in development for B-cell leukemias.22 Dual-targeting CARs or combination therapy may prevent relapses due to escape variants and therefore may be the way forward.
Conclusions
CD19-targeted CAR-modified T-cell therapy has proven CARs to be extremely powerful agents with unprecedented results in patients without curative options. Moreover, this highly active cell therapy has paved the way for CARs with diverse targets. Future work focusing on target identification and toxicity management will broaden the clinical applicability and bring this exciting therapy to more patients.
Disclosures
Conflict-of-interest disclosures: S.L.M. and E.J.S. declare no competing financial interests. S.A.G. has received research funding from and has consulted for Novartis. Off-label drug use: None disclosed.
Correspondence
Stephan A. Grupp, MD, PhD, 3006 Colket Translational Research Bldg., 3501 Civic Center Blvd., Philadelphia, PA 19104; Phone: (215)590-5476; Fax: (215)590-3770; e-mail: grupp@email.chop.edu.
