
Abstract
Cancer patients are at an increased risk of venous thromboembolism. The clotting system is activated in most cancer patients, which is reflected by specific parameters such as an increased thrombin generation and elevated D-dimer levels. Blood cells, especially WBCs and platelets, play an important role in this activation process. Neutrophils and monocytes are subpopulations of WBCs that increase the thrombotic potential by different mechanisms. Neutrophils are activated by tumor cells and can release DNA, generating highly thrombogenic neutrophil extracellular traps. Monocytes are able to synthesize and express significant quantities of procoagulant tissue factor on their surfaces upon activation. An increased risk of VTE has been found in patients with solid tumors and elevated platelet count and in those with high-grade gliomas and low platelet count. Small circulating membrane vesicles, also called microparticles (MPs), which largely derive from platelets, contribute to the procoagulant potential. Specifically, procoagulant MPs could play a role in tumor-associated thrombosis in pancreatic cancer. Interventional studies are under way that are investigating the benefits of thromboprophylaxis in patients identified to be at high risk of VTE through risk-scoring models that include blood count parameters. The “flames” thrown by blood cells, such as neutrophil extracellular traps and MPs, although exciting, still have to be investigated for their usefulness in the clinical setting.
Learning Objectives
To understand that blood cells play an important role in cancer-associated VTE
To understand that altered parameters of blood cell count have been reported to be associated with incident VTE; however, many still have to be validated in interventional studies
To understand that the “flames” thrown by blood cells, such as MPs and NETs, although exciting, still have to be investigated for their usefulness in the clinical setting
Introduction
Cancer is an important risk factor for venous thrombosis and pulmonary embolism, commonly known as venous thromboembolism (VTE). Moreover, additional risk factors such as surgery or immobilization occur more often in cancer patients than in the general population. Cancer treatment, either pharmacologic or by irradiation, is also known to increase the risk of VTE. The probability of developing VTE varies considerably among cancer patients depending on the tumor type, disease stage, treatment modalities, and patient-dependent risk factors.1 From cohort studies and interventional studies that investigated the benefits of primary thromboprophylaxis in cancer patients, we have learned that, in an unselected cancer population, the risk of VTE is elevated, but not high enough to justify mandatory thromboprophylaxis in each cancer patient.2-4 In recent years, research has focused on predictive parameters to identify patients at low or high risk of VTE during the course of their disease. Patients with a low risk would not be candidates for thromboprophylaxis, with the exception of specific risk situations such as surgery. Patients at high risk would most probably benefit from a primary thrombosis prophylaxis over a certain period of time.
Blood cells (WBCs, RBCs, and platelets) are known to be altered in patients with cancer, not only in response to anticancer treatment, but may already be present at cancer diagnosis. Furthermore, blood cells may induce hypercoagulability by several mechanisms. All blood cells, but primarily platelets, can produce small vesicles, so-called microparticles (MPs), upon activation or during apoptosis that exert procoagulant activity. WBCs have been described to produce neutrophil extracellular traps (NETs) that might promote the activation of the clotting system and lead to VTE.
WBCs
Leukocytosis as risk factor
It is well known that the WBC count is frequently elevated in patients with cancer. This is particularly the case in patients with lung cancer. Interestingly, Thomson et al could show that the percentage of neutrophils was higher in patients who had a malignancy compared with those with benign disease.5 Therefore, this readily available parameter might be used clinically for diagnostic discrimination. Khorana et al first described an association between an elevated WBC count and an increased risk of VTE (Table 1).34 In the Awareness of Neutropenia in Chemotherapy (ANC) Study Group Registry, patients with an elevated leukocyte count (>11 × G/L) had a 2-fold increased risk of developing VTE. The association with VTE was only seen with neutrophils and monocytes, not with lymphocytes.2 For this review, we performed an evaluation of blood cell parameters' association with the risk of VTE in the framework of the Vienna Cancer and Thrombosis Study (CATS), a prospective observational cohort study that includes patients with various cancer entities.6 We took a dataset of CATS that was used in a recent publication by Königsbrügge et al, but only included patients with solid tumors.7 In the continuous analysis, WBC count was found to be associated with VTE. An increase of 1 G/L in the WBC was associated with a 7% increase in the risk of VTE [risk ratio (RR) = 1.06; 95% confidence interval (CI): 1.02-1.10; Table 2]. When the cutoff was set at 11 G/L according to the Khorana score, the RR was 1.34 (95% CI: 0.70-2.55). In a recently published population-based study including 24 034 initially cancer-free patients who were followed up until the development of cancer and/or VTE, elevated baseline WBC count (defined as ≥8.6 G/L) preceding the diagnosis of cancer was associated with a 2.4-fold higher risk of VTE.8
Blood count parameters and the risk of VTE in cancer patients (summarizing data from prospective cohort studies)

§Baseline WBC counts preceding the diagnosis of cancer and cancer-associated VTE.
Evaluation of blood count parameters and their association with VTE in the Vienna Cancer and Thrombosis Cohort (N = 988 patients with solid tumors only)
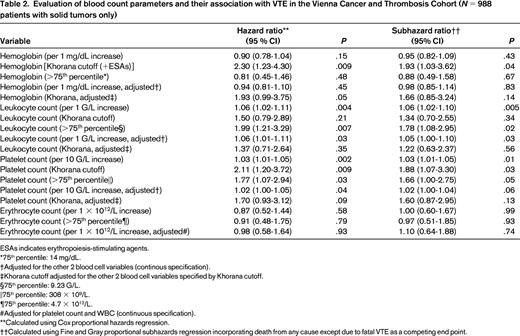
ESAs indicates erythropoiesis-stimulating agents.
*75th percentile: 14 mg/dL.
†Adjusted for the other 2 blood cell variables (continous specification).
‡Khorana cutoff adjusted for the other 2 blood cell variables specified by Khorana cutoff.
§75th percentile: 9.23 G/L.
‖75th percentile: 308 × 109/L.
¶75th percentile: 4.7 × 1012/L.
#Adjusted for platelet count and WBC (continuous specification).
**Calculated using Cox proportional hazards regression.
††Calculated using Fine and Gray proportional subhazards regression incorporating death from any cause except due to fatal VTE as a competing end point.
Leukocytosis also turned out to be associated with VTE in brain tumor patients. In a multivariable model adjusting for platelet count and D-dimer, the hazard ratio per double increase of WBC count was estimated to be 1.85 (95% CI: 0.99-3.41, P = .05).9
Neutrophils
A cellular “flame thrower” in the development of cancer-associated VTE appears to be the neutrophil granulocyte. Physiologically, neutrophils are the most abundant species of leukocyte and represent the main cellular component of the acute innate immune response to pathogens. Attracted by inflammatory cytokines, these highly motile cells rapidly migrate from the bloodstream to the site of infection, where they release aggressive antimicrobial agents and engage in phagocytosis of complement-tagged pathogens. In an additional independent mechanism, activated neutrophils combat infection by the release of DNA and histones, which subsequently form a dense local extracellular web that entraps bacteria and fosters the accumulation of microbicidal proteases called NETs. Importantly, NETs have been shown to be highly prothrombotic and act as a platform for further adhesion of platelets and erythrocytes.10
It is now well established that neutrophil activation occurs not only during infection, but also in inflammatory conditions such as cancer and autoimmunity independent of the presence of infection. In solid cancers, neutrophils represent a dominant cellular species in the local tumor-surrounding infiltrate.11 Elevated levels of circulating neutrophils (neutrophilia) are a characteristic feature of malignancy and are independently associated with a poor survival prognosis in a variety of human cancers.12
In terms of thrombosis in malignancy, neutrophilia per se has so far not been revealed as an independent risk factor for the development of VTE in cancer patients. Nevertheless, the recent discovery of highly thrombogenic NETs as key in vivo propagators of thrombosis in a murine cancer model supports the hypothesis that neutrophils may indeed represent a paramount causal factor for the prothrombotic state in malignancy.13
How could neutrophils cause thrombosis in patients with cancer?
Mechanistically, the concept of a “tumor-neutrophil-NETosis-thrombosis axis” appears to emerge from the very recent literature on this topic. Tumoral G-CSF overexpression and the presence of inflammatory cytokines such as Platelet Activating Factor (PAF) and IL-8 may represent the initial stimulus for priming a subset of neutrophils toward NET generation.13,14 These tumor-induced neutrophils are then activated via a still uncharacterized second hit to generate NETs (NETosis). Although the exact processes within the neutrophil that leads to chromatin degradation and subsequent NET secretion are not yet fully elucidated, it appears that the intracellular expression of Peptidylarginine Deiminase 4 (PAD4 or PADI4) is the key molecular step to NET formation.14 PAD4 citrullinates histones, which foster the degradation of chromatin and NET release. The mechanistic relevance of PAD4 for NET-induced thrombogenesis was elegantly shown in a murine inferior vena cava stenosis model by Demers et al, in which 9 of 10 PAD4 wild-type mice developed a thrombus within 48 hours after ligation, whereas only 1 of 11 PAD4-deficient mice did (Figure 1).13 Other molecules implicated in the intracellular NET formation pathway include neutrophil elastase (NE) and myeloperoxidase (MPO). NETs are then secreted into the extracellular space, where they form a dense prothrombotic web containing citrullinated histones, DNA, and histolytic enzymes such as NE, MPO, and cathepsin G. The individual components of this web may enter the circulation and exert local and systemic prothrombotic effects. Specifically, histones increase the generation of thrombin by impairing protein C activation16 and promote the release of VWF and P-selectin from endothelial Weibel-Palade bodies. Functionally, elevated VWF and thrombin may then lead to further platelet activation, whereas P-selectin sustains this process by attracting more leukocytes and platelets. Clinically, we have observed in the prospective Vienna CATS that biomarkers and tests such as an elevated in vivo and in vitro thrombin generation potential, VWF, and P-selectin, represent very strong and independent predictors of VTE in patients with cancer.4,17,18 In vitro, free DNA and RNA have been demonstrated to exert procoagulatory effects via activation of coagulation factors XI and XII. Finally, tissue factor (TF) colocalizes with NETs and NET-associated histolytic enzymes such as NE and cathepsin G degrade its antagonist TF pathway inhibitor directly, thus promoting coagulation via the extrinsic pathway.19

Evidence of in vivo NET formation in a murine cancer model. (A) The injection of tumor cells in mice increases the immunofluorescence of NETosis biomarkers DNA (blue) and citrullinated Histone 3 (H3) in murine neutrophils after stimulation with PAF; neutrophils from tumor-free mice show only minimal immunofluorescence for DNA and H3, whereas immunofluorescence increases after injection of the tumor (days 7 and 14). Scale bars indicate 20 μm in the top row and 5 μm in the bottom row. VWF- (V) and fibrin-rich thrombi form spontaneously in the lungs of tumor-bearing mice 28 days after tumor injection (n = 4); lung sections are immunostained for DNA (blue), VWF (green), and fibrinogen/fibrin (red). Scale bar indicates 50 μm. White star indicates a VWF- and fibrin-rich thrombus in the pulmonary vasculature. (C) Left, Tumor injection in mice increases the percentage of NET-generating neutrophils (as indicated by H3 hypercitrullination; plotted on the y-axis) from initially ∼1% in tumor-free mice to >80% at day 21 after tumor injection; the steep decline in the percentage of NET-generating neutrophils after day 21 was associated with spontaneous intravascular thrombus formation and may indicate a consumption of NETing neutrophils at the site of thrombosis. Right, Western blot analysis reveals the presence of H3 in the plasma of tumor-bearing mice 28 days after tumor injection. (Figure adapted with permission from Demers et al.13 )
Evidence of in vivo NET formation in a murine cancer model. (A) The injection of tumor cells in mice increases the immunofluorescence of NETosis biomarkers DNA (blue) and citrullinated Histone 3 (H3) in murine neutrophils after stimulation with PAF; neutrophils from tumor-free mice show only minimal immunofluorescence for DNA and H3, whereas immunofluorescence increases after injection of the tumor (days 7 and 14). Scale bars indicate 20 μm in the top row and 5 μm in the bottom row. VWF- (V) and fibrin-rich thrombi form spontaneously in the lungs of tumor-bearing mice 28 days after tumor injection (n = 4); lung sections are immunostained for DNA (blue), VWF (green), and fibrinogen/fibrin (red). Scale bar indicates 50 μm. White star indicates a VWF- and fibrin-rich thrombus in the pulmonary vasculature. (C) Left, Tumor injection in mice increases the percentage of NET-generating neutrophils (as indicated by H3 hypercitrullination; plotted on the y-axis) from initially ∼1% in tumor-free mice to >80% at day 21 after tumor injection; the steep decline in the percentage of NET-generating neutrophils after day 21 was associated with spontaneous intravascular thrombus formation and may indicate a consumption of NETing neutrophils at the site of thrombosis. Right, Western blot analysis reveals the presence of H3 in the plasma of tumor-bearing mice 28 days after tumor injection. (Figure adapted with permission from Demers et al.13 )
Can we exploit neutrophils for the prediction of VTE in patients with cancer?
The present mechanistic evidence indicates that neutrophilic NET formation could be the common denominator of cancer-associated VTE pathogenesis. However, the current enthusiasm about NETs in cancer is not yet supported by clinical data. Although 8 cancer patients were included in a case-control study on NET formation in thrombotic microangiopathy (N = 39)20 and Demers et al recently identified the presence of citrullinated histone3 in 3 cancer patients,14 specific evidence within a cancer population is lacking. Ideally, a causal relationship between NETosis and VTE in cancer would be established by demonstrating a temporal relationship between baseline NET specific biomarkers and the future occurrence of VTE in a large cancer population without clinically apparent VTE at baseline. Should this predictive relationship emerge in such a study, it is conceivable that NET biomarkers could be used clinically for the identification of cancer patients at high risk for VTE. Several NETosis candidate biomarkers can be imagined for this purpose, including, among others, circulating free DNA, citrullinated histone 3, NE, and MPO. Circulating free DNA is quantifiable with standard laboratory methods,20,21 and may be relatively specific for true NET-derived DNA. Citrullinated histone 3 appears to be very specific for PAD4-dependent neutrophilic chromatin release, but has so far only been measured qualitatively using Western blot.14 Finally, NE and MPO are exclusively derived from neutrophilic origin and would be readily quantifiable with commercial kits.
Synoptically, NETs appear to be the flames of neutrophils that set fire to the coagulation system in patients with cancer. Further significant developments on this topic may be expected in the near future when NET biomarkers will be assessed clinically as predictors of VTE in the cancer setting.
Monocytes
Circulating monocytes, which populate tissues as macrophages, represent a subgroup of leukocytes that are referred to as the mononuclear phagocyte system and play a determining role in innate and adaptive immunity. Monocytes are the only circulating blood cells that are able to synthesize and express significant quantities of highly procoagulant TF on their surface upon activation. TF is a transmembrane receptor and the main in vivo initiator of the blood coagulation cascade. A well-described mechanism of induction of TF expression on monocytes is the binding of the bacterial endotoxin lipopolysaccharide (LPS) to LPS-binding protein and CD14, which leads to the activation of the NF-κB signaling pathway.22 TF activity from LPS-stimulated monocytes was increased several-fold by the addition of platelets and granulocytes.23 This effect was suppressed by an antagonist to PAF and depended entirely on the P-selectin-PSGL-1 interaction. A substantial part of the TF activity was not found on the LPS-stimulated monocytes themselves, but on MPs in the supernatant (which will be discussed in a following paragraph). In a clinical study, Lwaleed et al found that monocyte TF expression was significantly higher in patients with urological carcinomas than in those with benign tumors and in healthy controls after in vitro stimulation with LPS.24 In another study that included patients with breast and colorectal cancer, it was demonstrated that levels of TF on LPS-stimulated monocytes were associated with tumor grade, stage, and the patients' survival time.25 Immunohistochemically, it was shown that tumor-associated macrophages, which are a major component of the infiltrate of tumors, express the coagulation factors II, V, VII, and X on their surface and therefore provide components for the extrinsic thrombin formation.26
At present, experimental data from studies investigating interactions between cancer cells and monocytes are still limited. Lindholm et al performed cell coculture experiments with monocytes and prostate cancer cell lines and found that monocytes stimulated TF expression on prostate cancer cell lines.27 In addition, it was shown that cocultivation of tumors with monocytes or macrophages led to increased tumor cell invasion and promoted factors involved in tumor aggressiveness.
Recently, Wang et al demonstrated that tumoral TF was a major source of coagulation activation in an orthotopic mouse xenograft model of human pancreatic cancer.28 Whether monocytes play a determining role in tumoral coagulation activation and thus also in promoting thrombosis via TF remains to be investigated.
RBCs
In many patients, the hemoglobin level is decreased at cancer diagnosis. This can be due to chronic blood loss, as in patients with gastrointestinal cancer, but is also found in other cancer patients and has been called “anemia of chronic disease.” In the ANC study group registry, Khorana et al described in a multivariable analysis that patients with cancer and a prechemotherapy hemoglobin <10 g/dL and/or the use of erythropoietin-stimulating agents had a 1.8-fold increased risk of developing VTE.2 In another prospective study by Mandala et al in patients with various cancer entities, hemoglobin levels could not be identified as a risk predictor for VTE (Table 1).29 The present evaluation of our CATS cohort revealed neither hemoglobin nor erythrocyte count as predictive parameters for cancer-associated VTE (Table 2).
Platelets
Platelets are the smallest hematopoietic cells, but are the ones most involved in hemostasis. On the site of vascular injury, platelets form a blood clot by adhesion and aggregation and promoting the activation of the clotting cascade that finally leads to the formation of fibrin. Activated platelets change their shape, release a large number of prothrombotic molecules from their α- and dense granules, and synthesize thromboxane A2. They also emit MPs and could thus be regarded as flame throwers for clotting activation. The role of the latter is separately described in this review. Platelets might not only play an important role in cancer-associated thrombosis, but may also influence the progression of cancer. The contribution of platelets to the pathophysiology of cancer and thrombosis has been reviewed recently by Riedl et al.30
In the ANC Study Group Registry, the rate of VTE was increased 2.8-fold in patients with a platelet count above the normal range (>350 × 109/L) before initiation of chemotherapy.2 Elevated platelet count as a risk factor for cancer-associated VTE was also investigated in a subset of 665 cancer patients with solid tumors within the Vienna CATS. In that study, the association between thrombocytosis and VTE could be confirmed, however, only when a higher cutoff level (95th percentile of the CATS cohort: >443 G/L) was applied.31 Patients with platelet counts above this cutoff had a 3.5-fold increased risk of VTE in multivariable analysis. For this review, we also evaluated the association of the platelet count with incident thrombosis in a more recent dataset of the CATS cohort in patients with solid tumors only (N = 988).7 Per 10 G/L increase in platelet count, the univariable RR was 1.03 (95% CI: 1.01-1.05). Using the Khorana-score cutoff of 350 G/L, the RR was 1.88 (95% CI: 1.07-3.30; Table 2). The cumulative probability of VTE after 1 year accounting for competing mortality was 18.7% in patients with a platelet count above our previously specified >443 G/L cutoff, as opposed to only 5.3% in those below (Figure 2).

Risk of VTE in the Vienna Cancer and Thrombosis Cohort according to baseline platelet count. Study included 988 patients with solid cancers only: breast (n = 197), lung (n = 213), gastric (n = 59), among others. High platelet count is associated with VTE in cancer patients. (Results shown are from Simanek et al.31 )
Risk of VTE in the Vienna Cancer and Thrombosis Cohort according to baseline platelet count. Study included 988 patients with solid cancers only: breast (n = 197), lung (n = 213), gastric (n = 59), among others. High platelet count is associated with VTE in cancer patients. (Results shown are from Simanek et al.31 )
Interestingly, when we evaluated the association between platelet numbers and occurrence of VTE more closely for different cancer entities, we observed that, in brain tumors, the relationship between platelet numbers and VTE was completely contrary to observations in solid tumors, as previously reported. In patients with high-grade gliomas, platelet count (as continuous variable per 50 × 109/L increase) was inversely associated with the occurrence of VTE (HR = 0.73, 95% CI: 0.53-0.95). This was a completely unexpected result and the underlying pathophysiological process has yet to be investigated. A score that included a low platelet count (<196 G/L), elevated D-dimer (>1.66 μg/mL), and an increased leukocyte count (>11.5 G/L) allowed us to discriminate between patients with a 2-year-VTE risk as high as 37.3% and patients with a corresponding VTE risk as low as 3.3%.9 It would be clinically important to identify predictors of VTE in this group of patients because patients with high-grade glioma are at a substantially increased risk of VTE; further, anticoagulation in high-grade glioma patients is associated with an increased risk of cerebral bleeding. A targeted thromboprophylaxis exclusively for patients with a high VTE risk would be preferable over routine prophylaxis in every patient.
Because there are several reports on the association of the size of the platelets with arterial or venous thrombosis, we recently investigated whether mean platelet volume (MPV) was associated with the risk of future VTE in the CATS cohort. Interestingly, we observed an association between small platelet size and increased risk of VTE, which was rather unexpected.32 High MPV (75th percentile of the study population; <10.8 fL) was associated with decreased risk of VTE compared with MPV below the 75th percentile (HR = 0.59, 95% CI: 0.37-0.95, P = .031). In multivariable analysis, including age, sex, cancer groups, newly diagnosed versus recurrent disease, platelet count, and soluble P-selectin, this association remained statistically significant. Specifically in patients with pancreatic cancer, the prognostic significance for VTE was impressive. In these patients, the HR of VTE in those with high MPV levels compared with patients with lower MPV was 0.15 (95% CI: 0.03-0.69, P = .02). The cumulative probability of developing VTE was 4.9% in patients with high MPV versus 20.2% in those with lower MPV after 6 months and 4.9% versus 33.4% after 2 years (Figure 3).

Risk of VTE in the Vienna Cancer and Thrombosis Cohort according to baseline MPV. The study included 99 patients with pancreatic cancer only with MPV below and above 10.8 fL. Competing risk analysis incorporating death from any cause except due to fatal VTE was the competing end point. (Figure adapted with permission from Riedl et al.32 )
Risk of VTE in the Vienna Cancer and Thrombosis Cohort according to baseline MPV. The study included 99 patients with pancreatic cancer only with MPV below and above 10.8 fL. Competing risk analysis incorporating death from any cause except due to fatal VTE was the competing end point. (Figure adapted with permission from Riedl et al.32 )
It is a relatively novel observation that platelets play an important role not only in arterial thrombosis but also in venous thrombosis, specifically in cancer-associated VTE. This finding is now being already implemented in the primary prevention of VTE by the use of aspirin as thromboprophylaxis in patients with multiple myeloma (MM). Patients with MM who are treated with thalidomide or lenalidomide in combination with chemotherapy and dexamethasone are at an increased risk of VTE. A 3-arm study showed that aspirin was as effective as low-molecular-weight heparin in the prevention of VTE in MM patients.33
The important role of platelets in cancer-associated thrombosis is further supported by the observation of a strong association between soluble P-selectin and VTE in cancer patients. In CATS, this association was seen in both the inception cohort and also in a validation cohort.4,6 The inclusion of soluble P-selectin together with D-dimer in a VTE risk-scoring model in addition to the variables previously suggested by Khorana34 improved the predictive performance for VTE in cancer patients.4
MPs
Cells that are activated or undergo apoptosis release procoagulant MPs (also referred to as microvesicles or extracellular vesicles) into the circulation. MPs are most commonly defined by their size (ranging between 0.1 and 1 μm) and the expression of a phospholipid called phosphatidylserine (PS) that is able to bind coagulation factors. Platelet-derived MPs are the most abundant MPs in plasma, but MPs also originate from erythrocytes, endothelial cells, lymphocytes, monocytes, smooth muscle cells, and cancer cells.35 MPs were reported to be 50- to 100-fold more prothrombotic than the equivalent area on activated platelets. Specifically, a substantial part of the procoagulant potential of blood was attributed to MPs. However, in a large group of prospectively followed cancer patients, levels of PS exposure on MPs, quantified with a prothrombinase assay, did not predict an increased propensity for future VTE.36
Experimental and clinical studies have shown that TF-bearing MPs represent a particularly procoagulant MP ubpopulation that is likely to play an important role in the prothrombotic state found in different malignancies. First, Dvorak et al noticed that TF-bearing MPs are spontaneously shed from different cancer cell lines into the cell culture medium. Injection of in vitro-generated TF-bearing MPs from a pancreatic cancer cell line into the tail vein of mice led to a strong activation of the coagulation system that was reversed by heparinization.37 Surprisingly, in 2 studies that used 2 orthotopic xenograft mouse models of human pancreatic cancer, a strong prothrombotic effect of in vivo-generated TF-bearing MPs could not be clearly demonstrated. In one of these studies Davila et al found no effect of in vivo generated TF-bearing MPs on fibrin clot formation.37 In the second study, Wang et al found elevated thrombin-antithrombin and D-dimer levels in mice containing orthotopic human pancreatic tumors. However, applying an inferior vena cava stenosis model, they detected similar-sized thrombi in mice with elevated tumor-derived MP-associated TF (MP-TF) activity and in controls.38 Consistent with these data, we demonstrated that TF-bearing MPs from pancreatic cancer patients only had a small but statistically significant effect on fibrin clot formation, which might be explained by the relatively low PS coexpression on these MPs compared with highly procoagulant monocyte-derived TF-bearing MPs.39
In a cross-sectional clinical study, Tesselaar et al found elevated MP-TF activity levels in 7 breast and pancreatic cancer patients with acute VTE compared with controls. The finding of elevated MP-TF activity in cancer patients with acute VTE compared with those without VTE was confirmed in a subsequent clinical study.40 Results from prospective studies that investigated MP-TF activity as potential biomarker for prediction of future VTE in cancer patients are inconsistent. In a prospective study that included patients with 4 highly prothrombotic cancer types, pancreatic, colon, gastric, and brain cancer, we found a borderline significant association between MP-TF activity and future VTE in pancreatic cancer patients only. Consistently, Auwerda et al, who included 122 patients with multiple myeloma, and Hernández et al, who included 252 patients with different cancer types, did not observe an association between MP-TF activity and future VTE.41,42 Conversely, in a case series of 11 pancreatic cancer patients, of whom consecutive blood samples were drawn, Khorana et al found that 2 patients with rising MP-TF levels developed VTE.45 Zwicker et al found an association between TF-bearing MPs and future VTE applying impedance-based flow cytometry in patients with different malignancies (N = 208).43
Synoptically, the current experimental data suggest that PS- and TF-bearing MPs play an important role in the pathogenesis of cancer-related VTE. However, the currently available clinical data do not indicate that TF-bearing MPs play a determining role as a biomarker for the prediction of cancer-related thrombosis. It needs to be considered that this could also be due to methodological limitations and/or a lack of large sufficiently powered prospective studies.
Acknowledgments
The authors thank Johannes Thaler for adding most valuable content, Cihan Ay for critical comments, and Tanja Altreiter for proofreading the manuscript.
Disclosures
Conflict-of-interest disclosures: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Prof. Ingrid Pabinger, MD, Medical University of Vienna, Department of Medicine I, Clinical Division of Haematology and Haemostaseology, Waehringer Guertel 18-20, A-1090 Vienna, Austria; Phone: 43-1-40400-44480; Fax: 43-1-40400-40300; e-mail: ingrid.pabinger@meduniwien.ac.at.
