
Abstract
Treatment of patients with hemophilia A and B has undergone significant advances during the past 2 decades. However, despite these advances, the development of antibodies that inhibit the function of infused clotting factor remains a major challenge and is considered the most significant complication of hemophilia treatment. This chapter reviews current tools available for the care of patients with inhibitors and highlights areas where progress is imminent or strongly needed. For management of bleeding, bypassing agents remain the mainstay of therapy. Recombinant factor VIIa and activated prothrombin complex concentrates are similarly effective in populations of patients with hemophilia and inhibitors; however, individuals may show a better response to one agent over another. Recent studies have shown that prophylaxis with bypassing agents can reduce bleeding episodes by ∼50%-80%. The prophylactic use of bypassing agents is an important tool to reduce morbidity in patients before they undergo immune tolerance induction (ITI) and in those with persistent high titer inhibitors, but cost and lack of convenience remain barriers. Because of the significant burden that inhibitors add to the individual patient and the health care system, inhibitor eradication should be pursued in as many patients as possible. ITI is an effective tool, particularly in patients with severe hemophilia A and good risk profiles, and leads to a return to a normal factor VIII response in ∼60% of patients. However, for the group of patients who fail to respond to ITI or have hemophilia B, new and improved tools are needed.
Learning Objectives
To be able to describe the benefits and general approach for preventing joint bleeding in patients with hemophilia and inhibitors
To be able to select the most appropriate ITI regimen given the patient's clinical inhibitor profile
Introduction
The development of neutralizing antibodies (inhibitors) to factor VIII (fVIII) or factor IX (fIX) is the most significant complication of hemophilia treatment, occurring in up to 33% of patients with severe hemophilia A, in 13% of those with nonsevere hemophilia A,1 and in 3% of patients with severe hemophilia B.2 In the presence of an inhibitor, the risks of major morbidity and the cost of care increase substantially. The consequences of bleeding and the demands of treatment increase the disease burden on patients and their families, leading to reduced quality of life, financial stress, and strained relationships.3,4 For these reasons, improved management of patients with hemophilia complicated by an inhibitor is an important, timely challenge for the health care community. This chapter reviews current tools for the care of patients with inhibitors, focusing on treatment and prevention of bleeding and inhibitor eradication. Areas in which progress is imminent or strongly needed are highlighted. Because the incidence and prevalence of inhibitors is higher in patients with severe hemophilia A and the body of literature evaluating the management of inhibitors in these patients is also larger, this chapter focuses on severe hemophilia A; the management of inhibitors in nonsevere hemophilia A and hemophilia B are discussed only briefly.
Treatment of bleeding
The major morbidity that results from the development of an inhibitor in patients with hemophilia is bleeding that is difficult to treat. Although hemostatic therapies that bypass the missing clotting factor are available, nothing works as well as replacement therapy with clotting factor concentrates. For this reason, in patients with low-responding inhibitors, continued treatment with concentrates at the same or higher doses is preferred. Once an inhibitor titer is >5 Bethesda units (BU)/mL, factor concentrates are typically ineffective and bypassing agents are used. As their name implies, bypassing agents treat bleeding by producing thrombin via pathways that do not require fVIII or fIX. Bypassing agents that are currently available include recombinant factor VIIa (rfVIIa, Novoseven RT; NovoNordisk) and activated prothrombin complex concentrates (aPCC, FEIBA VH; Baxter; Table 1).
Currently available bypassing agents for treatment and prevention of bleeding
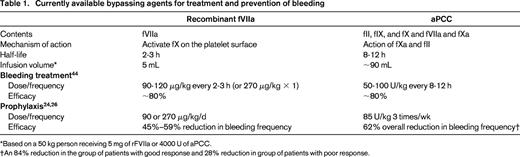
*Based on a 50 kg person receiving 5 mg of rFVIIa or 4000 U of aPCC.
†An 84% reduction in the group of patients with good response and 28% reduction in group of patients with poor response.
Both rfVIIa and aPCC have been studied across various clinical scenarios and have had an overall efficacy rate of >80% with similar rates of adverse events.5,6 The FENOC study compared these 2 agents in a randomized fashion, with control of joint bleeding 6 hours after initiating treatment as the primary outcome.7 There appeared to be similar hemostatic efficacy between the products in the treatment of joint bleeds, although the criterion of statistical equivalency was not met. However, in patients who received both products, ∼1/3 reported better efficacy with one product (rFVIIa or aPCC) over the other. Therefore, it is important to individualize bypass therapy. Other parameters that may drive choice include infusion requirements (higher volume but less frequent with aPCC) and the avoidance of allergic reactions and anamnesis, both of which can be seen with aPCC due to the presence of fIX and small amounts of fVIII.
Two major challenges in treating patients with inhibitors are predicting which product a patient will respond to and treating patients who have a poor hemostatic response to both agents. Both calibrated automated thrombin generation testing and thromboelastography, 2 investigational assays of global hemostasis, have been shown to predict the response to bypassing agents.8,9 A prospective study evaluating the hemostatic response of patients with inhibitors undergoing surgical procedures showed that in vitro selection of bypassing agent and dose based on thrombin generation correlated with a good clinical hemostatic response.10 Although these reports are promising, worldwide standardization of both the calibrated automated thrombogram and thromboelastography is needed before routine clinical use, so these procedures continue to be works in progress.11
For patients who have poor response to both rfVIIa and aPCC as single agents, combination therapy using both agents has been reported, but must be used with caution because of the increased risk of thrombosis.12 In a recent study, the combination of tranexamic acid and either rfVIIa or aPCC was shown to improve clot stability compared with a bypassing agent alone in a small number of patients with hemophilia and inhibitors.13 Other alternative therapies include porcine fVIII and high doses of human fVIII. Porcine fVIII is effective because antihuman fVIII antibodies show limited cross-reactivity with porcine fVIII. Recombinant porcine fVIII (rpfVIII, OBI-1) was shown to be effective in 28 patients with acquired fVIII inhibitors and serious bleeding.14 At this time, the use of rpfVIII remains investigational; however, according to a manufacturer (Baxter) press release on December 10, 2013, a biologics license application for use in acquired hemophilia A has been submitted to the US Food and Drug Administration (FDA). Its use in congenital hemophilia A complicated by an inhibitor will require additional investigation. The routine use of rpfVIII may be limited given the potential risk of developing antiporcine fVIII antibodies after repeated exposures. However, there have been reports of patients receiving plasma-derived porcine fVIII for extended periods of time without antibody cross-reactivity and with continued good clinical response.15 High-dose human fVIII is an option for selected patients with a high-responding inhibitor that has decreased to a titer <5 BU/mL at the time of a serious limb or life-threatening bleeding event. There have also been recent reports of improved thrombin generation ex vivo with combinations of fVIII and bypassing agents in samples from patients with hemophilia A and inhibitors and in vivo in a small pilot study.16,17 Response to fVIII may be dependent on the epitope specificity of the inhibitor, because nonclassical anti-C2 antibodies have been shown to be overcome by fVIII in an in vitro thrombin generation model and an in vivo murine tail-bleeding model; conversely, despite lower inhibitor titers, classical anti-C2 antibodies cannot. Knowledge of epitope specificity may facilitate selection of fVIII as a therapeutic option.18
The limitations of bypassing agents have spurred the development of both novel bypassing agents and longer-acting rfVIIa compounds. Unfortunately, the development of several promising agents has been halted due to adverse events; vatreptacog alfa due to development of antidrug antibodies in a few patients (one with neutralizing effect; Novo Nordisk press release dated August 9, 2012), the long-acting rfVIIa, BAY 86-6150 due to the formation of neutralizing antibodies (Bayer press release dated May 3, 2013), and anti-tissue factor pathway inhibitor aptamer (ARC 19499) due to increased bleeding in the phase 1/2 clinical trial.19 Other novel experimental agents in development include those that block tissue factor pathway inhibitor, an antifactor IXa/X-bispecific antibody, a zymogen-like factor Xa molecule, and an RNA interference (RNAi) therapeutic targeting antithrombin.20-23 Unlike current products that must be given IV, these new products may ultimately lead to adjunctive treatment for patients with hemophilia by subcutaneous or oral routes. Despite the failed development of some agents, the potential of new therapeutics to improve hemostasis in patients with an inhibitor remains promising.
Prevention of bleeding
As the use of prophylaxis in patients with hemophilia without inhibitors, even in the setting of preexisting joint disease, has become more routine and its benefits more apparent, there has been increasing interest in prophylaxis with bypassing agents to prevent bleeding episodes in patients with inhibitors.
Prophylaxis using aPCC
In the Pro-FEIBA study, patients >2 years of age with a high-responding inhibitor and frequent bleeding currently treated with bypass therapy were randomized in a crossover design to receive aPCC prophylaxis 85 U/kg 3 times weekly or on-demand treatment.24 Among the 26 subjects included in the per-protocol analysis, joint bleeding episodes were reduced from a mean of 10.8 to 4.2 per 6-month period. Interestingly, a poor response, as evidenced by minimal to no change in the frequency of bleeding epsiodes, was seen among 38% of the subjects. Those with a good response to aPCC also demonstrated improvements in health-related quality of life (HRQoL) as measured by the SF-36 in dimensions related to physical and social functioning and pain. In addition, days missed from work due to bleeding or infusions were significantly reduced during the prophylaxis period.25
Prophylaxis using rfVIIa
In a multicenter, randomized, double-blind parallel group study, 22 subjects with hemophilia A and a high-responding inhibitor currently treated with a bypassing agent were randomized to receive rfVIIa 90 or 270 μg/kg/d for a 3-month period.26 In this study, during the preprophylaxis period, 5.6 and 5.3 bleeds per month were reported in each group, respectively. During the 3-month prophylaxis period, this was reduced to 3.0 bleeds per month in the 90 μg/kg group and 2.2 bleeds per month in the 270 μg/kg group. Target joint bleeds were reduced 43% and 61% in the 90 and 270 μg/kg groups, respectively. HRQoL also improved, although not statistically significantly. Reductions in hospitalizations and days missed from school or work were also observed.27
Prophylaxis during immune tolerance induction
aPCCs have long been used in conjunction with immune tolerance induction (ITI) and was part of the original Bonn protocol. Currently, the use of rfVIIa or aPCC for prophylaxis during ITI is typically reserved for patients with bleeding despite a high-dose ITI regimen and an inhibitor titer after the initiation of ITI that is >10 BU/mL. Once the inhibitor titer drops to <10 BU/mL or when there is measurable factor recovery, the clinical indication for prophylaxis should be reevaluated as the risk of catheter-associated thrombosis increases.28 The risk of thrombosis with ITI combined with rfVIIa or aPCC prophylaxis in the absence of a central venous access device is not well defined, so a clinically individualized approach balancing the potential risk of thrombosis with the need for prevention of bleeding is required.
Choice of bypassing agent
Both aPCC and rfVIIa can reduce the frequency of bleeding episodes. The choice of agent will depend on the current clinical situation, efficacy for the individual patient, and convenience. Because aPCC contains a small amount of fVIII that may lead to a rise in the inhibitor titer, its use is often avoided before the initiation of ITI. Given the lack of efficacy to a bypassing agent seen in some patients in both the Pro-FEIBA study and the FENOC study, a patient's responsiveness to a specific bypassing agent needs to be assessed and their regimen for prophylaxis tailored.7,24 Lastly, daily treatment with rfVIIa, necessitated by a shorter half-life, is difficult and costly when used for extended periods, favoring aPCC over rfVIIa for routine prophylaxis in hemophilia patients with an inhibitor.
Although prophylaxis using bypassing agents can reduce bleeding frequency and improve measures of health care utilization and HRQoL, subjects in these studies continued to have bleeding at a significant frequency and greater than would be anticipated in persons with hemophilia A or B receiving factor concentrates for prophylaxis. The limited success of prophylaxis with bypassing agents in patients with inhibitors highlights the need for new tools to prevent bleeding episodes that are effective and easy to administer.
Immune Tolerance Induction
ITI refers to the regular, frequent, and prolonged exposure of the patient to clotting factor with the goal of inducing peripheral tolerance. ITI is currently the mainstay of treatment to eradicate an inhibitor, particularly in patients with severe hemophilia A (Figure 1). Possible mechanisms by which tolerance is induced by ITI include inhibition of B-cell memory and induction of T-cell anergy, anti-idiotypic antibodies, or suppressor T cells.29 Overall, ITI is successful in ∼70% of patients with hemophilia A and 30% of those with hemophilia B.30 When successful, patients can return to using factor concentrates for the routine prevention and treatment of bleeding. Despite the overall success of ITI, several challenges remain.

Proposed algorithm for ITI in patients with severe hemophilia A. CVAD indicates central venous access device; and PD, plasma-derived.
Proposed algorithm for ITI in patients with severe hemophilia A. CVAD indicates central venous access device; and PD, plasma-derived.
Severe hemophilia A
Patient selection.
In the setting of a newly identified inhibitor in a patient with hemophilia, the first consideration for ITI initiation is whether the inhibitor is high responding (≥5 BU/mL) or low responding (<5 BU/mL). Overall, ∼25% are low-responding inhibitors; some of these will be transient, resolving within 6 months. For patients with a persistent low-responding inhibitor, factor replacement therapy can often be used successfully for the prevention and treatment of bleeding with modestly increased doses. Once prevention and treatment of bleeding becomes less predictable and more difficult with factor replacement therapy, ITI should be considered. For patients with an inhibitor titer between 5 and 10 BU/mL, ITI should be started as soon as possible. For those with an inhibitor titer >10 BU/mL in the absence of severe bleeding, it has traditionally been recommended to delay starting ITI until the inhibitor titer is <10 BU/mL, preferably within 2 years of inhibitor onset.28 More recently, Nakar et al reported that successful tolerance (defined in this study as a negative inhibitor titer and the ability to use fVIII for the treatment of bleeding episodes) was achieved in 13 patients with severe hemophilia A who started ITI within 1 month of inhibitor detection despite pre-ITI titers >10 BU/mL (median 25 BU/mL, range 265 BU/mL), raising doubt as to the necessity of delaying ITI.31
In patients with high-responding inhibitors, there have been several predictors of success identified in registries and cohort studies and used in subsequent clinical trials of ITI (Table 2).32-34 Race has been proposed as a risk factor, but its independent effect is unclear.35 From these studies, a possible profile of good and poor risk has emerged (Table 3).

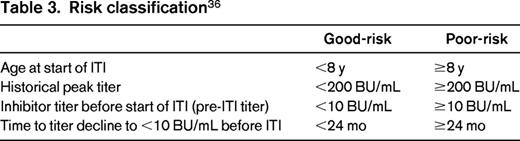
Based on the data from the International ITI study (IITI), in which 69.7% of subjects were rendered tolerant after 14-16 months of treatment, ITI is considered standard of care in patients with hemophilia A and high-responding inhibitors with good risk features.36 Providers caring for patients with high-responding inhibitors that do not fit within the good risk category defined by the IITI study, and therefore considered poor risk, must rely on observational data to determine the likelihood of successful ITI. In a report of 9 patients treated at a single center where ITI was started more than 2 years after inhibitor onset (regimens ranged from 50 IU/kg 3 times per week to 100 IU/kg daily), 4 of 9 subjects were successfully made tolerant and 3 more partially tolerant (inhibitor titer <5 BU/mL and able to prevent and treat bleeding episodes with fVIII). Five of these 7 had at least 1 additional risk factor for poor ITI response (pre-ITI titer >10 BU/mL and/or historical peak titer >200 BU/mL).37 In the International Immune Tolerance Registry (IITR), ∼40% of patients were successfully made tolerant in the setting of one of the following characteristics: age >20 years, peak inhibitor titer greater than 500 BU/mL, or pre-ITI titer >20 BU/mL.34 These reports suggest that ITI can be successful in poor-risk patients, albeit at a lower rate than in those with a good risk profile.
In these poor-risk patients, the likelihood of success and longer-term benefit of ITI needs to be weighed against the risks and costs measured in both dollars and time associated with ITI therapy. In the National Immune Tolerance Registry (NAITR), adverse events were unlikely in patients with hemophilia A, occurring in only 6% of patients.33 As the use of prophylaxis in patients with severe hemophilia becomes routine, the cost associated with ITI is limited to the period before tolerance is achieved and the dose of factor replacement tapered. In the absence of improved therapies, better models to predict the probability of successful ITI in an individual patient given a set of known risk factors before the start of ITI are needed, along with decision models that weigh the probability of success with the cost and risk.
Dose.
The optimal dose for ITI has long been debated. The debate was fueled by observations that low doses were associated with greater success in the NAITR and higher doses in the IITR.33,34 To solve this problem, the IITI study was designed and completed. The study demonstrated that, in good-risk patients, the probability of successful tolerance was similar (although not statistically equivalent) using a low-dose (50 IU/kg 3 times per week) and a high-dose (200 IU/kg/d) regimen.36 However, despite the similar success rates, the time to success varied. Importantly, the time to a negative inhibitor titer was 9.2 months [interquartile range (IQR) 12.1] in the low-dose arm and 4.6 months (IQR 11) in the high-dose arm. This translated to a significantly greater risk of bleeding in the low-dose arm during this early phase (hazard ratio = 2.27; 95% confidence interval, 1.29-4.01). Based on these results, higher doses are favored over lower doses. However, regimens can be adapted based on the patient's bleeding frequency and quality of venous access. Ultimately, an economic analysis of the IITI study will yield valuable insight into how to take cost into account when choosing between the 2 regimens without compromising patient outcomes.
In poor-risk patients, there is limited evidence upon which to base a recommendation of one regimen over another. In the meta-analysis of the IITR and NAITR, it was observed that patients with historical peak titers >200 BU/mL and /or pre-ITI titers of >20 BU/mL had a higher probability of achieving tolerance with a high-dose regimen (200 IU/kg/d) than when lower doses were used.38 From these observations and those of the IITI study during the first phase, when bleeding risk was greatest (from the start of ITI until a negative inhibitor titer), higher dose regimens are favored. However, some patients, particularly adults in whom poor risk features are more common, are unable or unwilling to do daily high-dose ITI. In many of these patients, it is our opinion that it is still preferable to undergo a trial of ITI using a regimen that is feasible for the patient rather than to abandon ITI altogether.
Product choice.
The type of product to use for ITI has been a matter of debate since the observation by several German treatment centers that ITI success rates declined after switching from plasma-derived fVIII to monoclonal purified or recombinant fVIII products.39 Although these reports are provocative, they were limited by a lack of control for other confounding factors. A meta-analysis of 13 studies involving 382 patients did not support an association between product type and outcome; however, information on pre-ITI risk (good vs poor) was incomplete, thereby limiting confidence that the groups were similar.40 This question will likely remain a matter of debate for some time because a prospective, randomized controlled trial of ITI comparing FVIII concentrates with and without VWF in patients with poor-risk features undergoing a first course of ITI was closed due to poor accrual. The RES.I.S.TExperience study remains open for accrual, but this is a single-arm, open label study of ITI using VWF-containing products in patients who had previously failed ITI.41 This study's target enrollment is 50 patients and will provide useful information about the effectiveness of VWF-containing products for ITI in patients who previously failed ITI. Unfortunately, in the absence of a comparison with a second course of ITI using non-VWF-containing concentrate, the relative benefit of a VWF-containing product in those with a prior history of ITI failure will remain unknown.
With the introduction of fVIII products that are fused to an Fc protein or albumin or are glycopegylated, questions arise as to their impact on initial tolerance and ITI. Although no trials investigating the use of these products in either previously untreated patients or patients with preexisting inhibitors have been completed, it has been speculated that these products may be less immunogenic. Mechanisms for the reduced immunogenicity include reduced binding capacity of glycopegylated fVIII to human-monocyte-derived dendritic cells42 and an increase in FoxP3-positive T-regulatory cells by IgG-Fc-containing peptides.43 Whether these longer-acting products have reduced immunogenicity or will provide an advantage over traditional fVIII products in the induction of peripheral immune tolerance after initial inhibitor development remains to be seen.
Immunosuppression during ITI.
Given that an unwanted immune response to fVIII underlies the development of an inhibitor, it follows that medications that modulate the immune system may facilitate inhibitor eradication. Along these lines, cyclophosphamide was used in the original Malmo protocols and was administered on days 1-3, followed by IVIg. In contrast, the NAITR failed to show any benefit of immunosuppressive medications, although the small sample size limits the power of this comparison.33 Because many patients, particularly those who are good risk, are able to be successfully rendered tolerant with factor concentrates alone, the use of immunosuppressive medications is currently not considered a routine part of ITI in patients with hemophilia A.28,44 However, there remains a fraction of patients with poor-risk features who are ITI naive or have failed a maximized regimen of ITI despite an initial good risk, for whom medications to modify the immune system could be considered.
Rituximab, an anti-CD20 antibody with a generally favorable safety profile, is often considered first among possible immunosuppressive medications. Most of the data for rituximab is derived from case reports and case series. In these reports, the overall response rate in patients with severe hemophilia is reported to be 40%–50% when rituximab is used concomitantly with ITI, although durable remissions occur in only a fraction of those with an initial response.45 Recently, a prospective, open-label, single-arm study of rituximab without concomitant ITI in patients with severe hemophilia and high-responding inhibitors (Rituximab to Treat Severe Hemophilia A, RICH study) was completed. Among the 23 subjects enrolled, 16 were challenged with fVIII, had an increase in their inhibitor titer to ≥5 BU/mL, and went on to receive rituximab. A major response was defined as an inhibitor titer <5 BU/mL and lack of anamnesis after rechallenge with fVIII, whereas a titer that was between 5 and 10 BU/mL but still less than 50% of the original anamnestic peak defined a minor response. A major response was seen in 18.8% (3/16) and a minor response in 6.2% (1/16).46
Determining ITI outcome.
Successful ITI has been defined by consensus groups and similar definitions have been used in clinical trials (I-ITI).28,36 Broadly, tolerance to fVIII is demonstrated when an inhibitor is no longer detected (negative Bethesda assay) and a normal pharmacokinetic response to fVIII infusion is observed. A recovery of 66% of expected and a half-life of >6 hours have been considered sufficiently normal pharmacokinetic responses to characterize complete tolerance, although some have argued that a longer half-life (>7 hours) should be the goal. Partial tolerance is typically defined as an inhibitor titer <5 BU/mL and the ability to use fVIII to prevent and treat bleeding despite a recovery <66% and/or half-life <6 hours. Failure of tolerance, the absence of partial or complete tolerance, can be more difficult to identify. In general, the absence of a reduction in bleeding episodes, the lack of a 20% decrease in inhibitor titer over a 3- to 6-month period, or an inhibitor titer >5 BU/mL after 3-5 years of ITI are markers of failure.36,47
Risk of recurrence.
Data on long-term follow-up after successful ITI have been limited. In the 1-year follow-up of the IITI study, 6 subjects demonstrated evidence of relapse at a median of 9.5 months (IQR 9 months), 1 with a measurable inhibitor titer and 5 with reduced fVIII recovery.36 In the NAITR, 48 of 75 subjects remained tolerant while on fVIII maintenance therapy with a median follow-up of 13 months (range 129).48 Nine subjects had a recurrent inhibitor, 3 after cessation of maintenance therapy and 6 while receiving maintenance therapy. In the IITR, 6 of 128 (4.7%) relapsed between 1 and 15 years. In the PROFIT registry, only 1 subject had a relapse after 7 years with a median study follow-up of 52 months.32 In another retrospective cohort study of patients who were successfully made tolerant at 12 US treatment centers with a median follow-up time of 4.3 years (IQR 5.6), the probability of any inhibitor recurrence was 0.15 at 1 year, 0.30 at 3 years, and 0.35 at 5 years.49 Clinical or treatment characteristics that influence inhibitor recurrence are unknown. As the numbers of persons that are successfully rendered tolerant grows, it will become increasingly important to understand the risk of inhibitor recurrence and the requirements to maintain tolerance.
Nonsevere hemophilia A
Inhibitors occur in those with mild or moderate hemophilia A less commonly than in those with severe disease; however, they pose a significant clinical challenge as the bleeding pattern of the patient often becomes more severe. In this population, the benefit of ITI is less clear. In a series of 36 cases of mild or moderate hemophilia A complicated by an inhibitor, multivariable analysis did not demonstrate any benefit of ITI.50 This observation is consistent with prior poor responses reported by Hay et al.51 Rituximab has been used more commonly in this group and, in contrast to severe hemophilia A, those with mild/moderate hemophilia A appear to have a better and more durable overall response rate; however, how this compares to observation alone in this population remains unknown.50
Hemophilia B
Given the lower prevalence of hemophilia B and the lower risk of inhibitor development in this group, the literature to support decisions regarding inhibitor eradication in patients with hemophilia B is scant. As occurs in patients with severe hemophilia A, transient low titer inhibitors can also occur in patients with hemophilia B.52 Accordingly, treatment with fIX can continue in the setting of a low titer inhibitor and adequate clinical response and the absence of allergic symptoms. ITI should be considered in those with a high-titer inhibitor or inadequate clinical response to fIX infusions (Figure 2). Overall, ITI is less effective, with a success rate of 31% in the NAITR.33 In addition, ITI is associated with a high risk of adverse events, including allergic reactions in 69% (11/16) and nephrotic syndrome in 19% (3/16).33 Other than allergic reactions, there is an absence of data to estimate the likelihood of success with ITI in an individual hemophilia B patient.2 Doses used have ranged from 25 to 200 IU/kg daily.33 There is insufficient information to associate a specific regimen (dose or product type) with outcomes. In patients with a history of allergic reaction to fIX, ITI can be cautiously considered after desensitization and in conjunction with immunosuppression; however, this is based on a limited number of case reports and case series and increases the burden of ITI.53,54 In patients without a history of allergic reactions, ITI should be more strongly considered. The use of immunosuppression in patients without a history of allergic reactions is not a part of routine practice, but given poor response rates to ITI alone, immunosuppression may be used during ITI after careful discussion with patients and their families. Immunosuppression regimens used have included the combination of IVIg, rituximab, dexamethasone, and mycophenolate mofetil.53 Monitoring for nephrotic syndrome during ITI is essential.

Gene therapy to promote immune tolerance
Gene therapy for the treatment of hemophilia has seen major advances in the past 5 years.55 The bleeding disorders community is hopeful that gene therapy for hemophilia B will become a therapeutic option in the near future, with hemophilia A to follow. With the observation of sustained plasma fIX levels after gene therapy, attention has turned to using gene therapy for induction of immune tolerance. Support for this therapeutic approach has been shown in the murine hemophilia B model in which liver-directed gene transfer using either adeno-associated viral vectors or lentiviral vectors can reverse preexisting anti-fIX high-titer inhibitors via T-regulatory cell induction.56,57 In a canine hemophilia A model, liver-directed gene therapy led to tolerance in 3 of 4 dogs.58 In addition, transplantation of hematopoietic stem cells engineered to express fVIII has been shown to induce fVIII-specific tolerance and eradicated fVIII inhibitors in mouse models.59,60
Conclusion
The development of an inhibitor is a major challenge to both patients and health care providers. For many patients with an inhibitor, bypassing agents can provide adequate hemostasis and prevent bleeding when used prophylactically; however, a subset of patients remain poor responders to both aPCC and rfVIIa. New tools to promote hemostasis for the prevention and treatment of bleeding are needed and new agents are in the pipeline. As a result of the challenges associated with the prevention and treatment of bleeding using bypassing agents, ITI should be considered in the majority of, if not all, patients with severe hemophilia A complicated by an inhibitor. Many good-risk patients will become tolerant with either low-dose or high-dose ITI and can continue on routine doses of factor concentrates after tolerance is achieved. For patients with hemophilia B, nonsevere hemophilia A, and severe hemophilia A and poor-risk features, ITI is less successful. The best strategy in these patients remains an open question.
This article was selected by the Blood and Hematology 2014 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2014. It is reprinted with permission from Blood 2014, Volume 124.
Disclosures
Conflict-of-interest disclosure: C.L.K. receives honoraria or serves on the board of directors or advisory committees for Baxter Healthcare, CSL Behring, and Kedrion Biopharma and receives research funding from Novo Nordisk. S.L.M. serves as a consultant for CSL Behring and receives honoraria or serves on advisory committees for Bayer Healthcare, Baxter Healthcare, and Grifols.
Correspondence
Christine L. Kempton, 1760 Haygood Drive, Health Sciences Research Building, Suite 340, Atlanta, GA 30322; Phone: (404)727-2846; Fax: (404)727-3681; e-mail: christine.kempton@emory.edu.
