
Abstract
Myeloproliferative neoplasms, including polycythemia vera (PV), essential thrombocythemia, and myelofibrosis (MF) (both primary and secondary), are recognized for their burdensome symptom profiles, life-threatening complications, and risk of progression to acute leukemia. Recent advancements in our ability to diagnose and prognosticate these clonal malignancies have paralleled the development of MPN-targeted therapies that have had a significant impact on disease burden and quality of life. Ruxolitinib has shown success in alleviating the symptomatic burden, reducing splenomegaly and improving quality of life in patients with MF. The role and clinical expectations of JAK2 inhibition continues to expand to a variety of investigational arenas. Clinical trials for patients with MF focus on new JAK inhibitors with potentially less myelosuppression (pacritinib) or even activity for anemia (momelotinib). Further efforts focus on combination trials (including a JAK inhibitor base) or targeting new pathways (ie, telomerase). Similarly, therapy for PV continues to evolve with phase 3 trials investigating optimal frontline therapy (hydroxyurea or IFN) and second-line therapy for hydroxyurea-refractory or intolerant PV with JAK inhibitors. In this chapter, we review the evolving data and role of JAK inhibition (alone or in combination) in the management of patients with MPNs.
Learning Objectives
To recognize the symptomatic burden present in lower-risk MPN populations and the role that targeted treatments may play in addressing this
To review the spectrum of available MPN-targeted treatments and the data involving their synergistic use with conventional treatments
Introduction
The classic BCR-ABL1-negative myeloproliferative neoplasms (MPNs) include polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis [both primary (PMF) and secondary (post-ET/PV MF)].1 ET and PV are characterized by increased platelet counts and erythrocyte counts, respectively, a heightened risk of thrombotic disease, microvascular symptoms, and survival rates that can approach those of the general population. MF, however, is traditionally a more problematic neoplasm with a frequently debilitating symptom profile that includes constitutional symptoms, splenomegaly, cytopenias, extramedullary hematopoiesis, risk of leukemic transformation (ie, MPN blast phase), and decreased life expectancy.2 Given the heterogeneous clinical needs of MPN patients, determination of when to treat, which agents to use, and how best to use them is often difficult. In this chapter, we review the recent advances in assessing symptoms and prognosis, as well as risk-based treatment strategies.
When to treat?
New MPN molecular insights
Management of patients with MPNs has evolved concurrently with our understanding of the molecular pathogenesis of these disorders. In 2005, the JAK2V617F mutation was found to be present in 95% of PV patients, 65% of PMF patients, and 55% of ET patients.3 A variety of other mutations such as JAK2 exon 12, MPL, IZF1, TP53, TET2, ASXL1, IDH1/2, and EZH2, also are believed to play a role in the pathogenesis of MPN disorders, with some potentially facilitating clonal section and promoting dominance of the JAK2V617F subclone.4-9 Many of these genes function in epigenetic regulation and often precede the JAK2 mutation, suggesting that the development of MPN disorders is the result of combined genetic deregulation as opposed to a single-hit mutation. In 2013, mutations in CALR (calreticulin) were reported to be prevalent in ET and PMF patients with nonmutated JAK2 or MPL.10,11 In a large study of 274 PMF patients, CALR mutated patients had improved overall survival at a median of 20.2 years compared with 8.2 years in wild-type patients.12 Recently, CALR-ASXL1+ mutational status was noted to be an important risk factor for inferior survival in PMF (HR 3.7).13 Whether the survival benefit is restricted to specific CALR subclones remains under investigation.14 In ET and PV, the CALR mutation is also associated with a decreased incidence of thrombosis and leukemic transformation.15,16 Data are emerging suggesting that CALR mutated patients may respond in a uniquely positive manner to therapies, particularly ET patients receiving IFN-α.17 In general, triple-negative patients (JAK2V617F, CALR, MPL) demonstrate inferior survival compared with those carrying the mutations.18 Despite our advances, the true role of molecular status in choice of MPN therapy is evolving and requires further validation before routine application.
MPN symptom burden
MPN patients can be highly symptomatic regardless of their subset of disease. Evidence supporting the role of symptoms in the diminution of both quality of life and life expectancy is substantial.19 MPN-specific patient-reported outcome tools (MPN-SAF20 and MPN 1021 ) have identified fatigue (87%), concentration problems (62%), early satiety (61%), inactivity (61%), night sweats (53%), itching (53%), abdominal discomfort (52%), bone pain (48%), weight loss (34%), and fevers (19%) to be prevalent and inadequately managed disease attributes (Figure 1).20,21 Symptoms are prominent in low-risk MPN populations as well. In a recent study assessing symptom burden response thresholds, it was noted that low-risk MF [Dynamic International Prognostic Scoring System (DIPSS)], PV (Tefferi criteria) and ET [International Prognostic Score for Essential Thrombocythemia (IPSET)] patients were moderately to highly symptomatic in 44%, 50%, and 41% of cases, respectively.22

MPN prognostic assessment
As stated, disease complications are a leading source of morbidity and mortality within the MPN disorders and play an important role in determining when to initiate therapy. Major thrombotic events range from 10% to 29% for ET and from 34% to 39% for PV.23 Hemorrhagic events are slightly less prevalent and both issues contribute to premature mortality. PV and ET remain at risk for transformation to MF and ensue a similar course to PMF in terms of symptoms, disease complications, and prognosis.24 Transformation to acute myelogenous leukemia occurs most frequently in MF, followed by PV and ET. Once acute myelogenous leukemia conversion has occurred, the median survival is 2.6-5 months.25 Prognostic tools have been developed for each of the MPN subtypes (Table 1). In ET, the IPSET has been validated to predict survival and the occurrence of thrombosis.26 For PV, the prognostic algorithm developed by Tefferi et al in 2013 remains the most widely used.27 For MF, most of the data exist for prognosis for those with PMF. Prognosis may be estimated using the International Prognostic Scoring System (IPSS) at the time of diagnosis or the DIPPS at any time during the disease process.28,29 An updated DIPSS-PLUS algorithm incorporates other independent risk factors, including RBC transfusion requirements, thrombocytopenia, and unfavorable karyotypes.2 Insights into the prognostic impact of CALR-ASXL+ status will likely result in their inclusion within DIPSS-PLUS assessment. Recognizing that symptom burden frequently contrasts that expected for a patients given risk status, a recent study explored symptom heterogeneity between risk score categories in MPN disorders (Figure 2A,B). A detailed analysis of this heterogeneity identified that unique phenotypic clusters are present within MPN disorders and may not be superimposed as surrogates for prognostic evaluations.30 The study also demonstrated that significant symptom burden is present within lower-risk MPN populations, who until recently have only been candidates only for conventional, nontargeted treatments.
MPN complication rates, prognosis and risk scoring algorithms
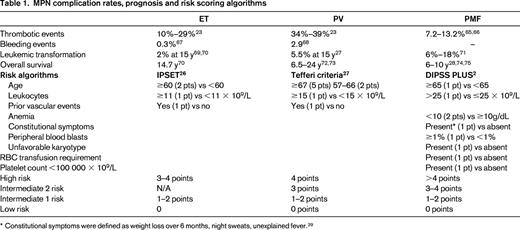
* Constitutional symptoms were defined as weight loss over 6 months, night sweats, unexplained fever.29

Current guidelines for timing of MPN therapy
The 2011 European Leukemia Network (ELN) treatment recommendations were among the first evidence-based MPN treatment algorithms to address the timeline and order of therapeutic interventions.31 Recognizing that the life expectancy in ET and PV may not be diminished in select populations, the primary goals of therapy for both PV and ET include prevention of thrombotic and bleeding complications in addition to minimizing the risk of progression to MF or acute leukemia. In contrast, MF treatment goals are based on assessment of both disease burden (symptoms, cytopenias, splenomegaly) and impact of disease on survival. It is important that the physician discuss the short- and long-term goals with the patient, covering potential sources of morbidity and impact on life expectancy. Details regarding the selection of appropriate therapies are discussed in the sections below.
Which agents to use
After discovery of the JAK2 mutation, numerous targeted therapies entered testing for MPN patients. Treatment options for MPN patients can be best divided into the categories of observation, medical therapies, and allogeneic stem cell transplantation (allo-SCT). Medical therapies themselves fall into the categories of cytoreductive agents, single-agent JAK inhibitors, combination approaches with a JAK inhibitor base, and non-JAK-targeted agents. Successful management of MPN patients along their disease course may use several of these approaches alone or in sequence.
Allo-SCT for MPNs
Allo-SCT is the only curative option for MF patients and is typically reserved for patients whose life expectancy is <5 years. This includes DIPSS intermediate-2 and high-risk individuals, along with patients with unfavorable cytogenetic abnormalities who are considered appropriate candidates in terms of their comorbidities and physiologic age.31 Overall survival is 50% for patients receiving conventional-intensity conditioning and life expectancy may be less than that expected if the patient did not undergo transplantation. Prospective, randomized studies specifically investigating features of the optimal allo-SCT candidate are lacking. Given the risks of graft failure, GVHD, and regimen-related toxicities associated with the procedure, physicians must consider the numerous factors that may affect outcome, including a patient's baseline RBC transfusion load, age, HLA donor status, and availability. There is a great deal of complexity associated with allo-SCTs in MF. Therefore, management of the disease begins with placing patients in a category of “urgent,” “delayed,” or “never” in regard to the timing of allo-SCT. Whether new therapies such as JAK2 inhibitors affect decision making for allo-SCT remains to be seen, but might first affect those in whom allo-SCT has already been selected.
Aspirin, phlebotomy, and cytoreductive medicine for MPN management
Low-dose aspirin for the prevention of vascular events has been evaluated in both PV and ET. For PV patients, the ECLAP study clearly demonstrated a reduced risk of vascular events without significantly increasing bleeding risk.32 Therefore, it remains frontline therapy for all PV patients regardless of risk score provided there are no absolute contraindications. The role of aspirin in ET is less clear in low-risk patients outside of the presence of microvascular symptoms. Evidence supporting the role of phlebotomy in all PV patients with hematocrit levels >45% was proven in the CYTO-PV trial, in which patients with controlled hematocrit had significantly reduced cardiovascular deaths or major thrombosis compared with more liberal hematocrit levels.33
Cytoreductive therapy is frequently used in high-risk PV and ET patients to reduce the occurrence of thrombohemorrhagic events, and treatment decisions are based on ELN recommendations.31 For both ET and PV, frontline cytoreductive treatment may include hydroxyurea (HU), which is proven to lower thrombotic complications. Anagrelide is a viable second-line therapy in ET. In the PT-1 trial, HU's superiority over anagrelide was demonstrated in in 809 ET patients at high risk for vascular events.34 Patients in the anagrelide group were significantly more likely to develop arterial thrombosis, serious hemorrhaging, and to have transformation to MF with a decreased risk of venous thromboembolism. Conversely, the ANAHYDRET study evaluating 259 high-risk ET patients compared anagrelide with HU (with the notable deletion of aspirin from the treatment plan) and did not identify any significant differences between groups in terms of arterial or venous thrombosis, severe bleeding events, or rates of discontinuation.35
In PV, nonleukemogenic pegylated (PEG) IFN-2α (Pegasys) is an acceptable alternative to HU and has been documented to induce complete hematological response in 76% and 77% of patients with PV and ET, respectively, and to result in decreases in JAK2 allele burden.36 Genotypic contexts were found to influence clinical and molecular responses. Currently under way is a randomized, open-label trial through the MPD Research Consortium evaluating the safety, toxicity, and tolerability of PEG-IFN-2α against HU in high-risk PV and ET patients (www.ClinicalTrials.gov identifier #NCT01259856). A phase 3 randomized, controlled study comparing the efficacy and safety of the novel monopegylated IFN-2β against HU in high-risk PV is also under way (PROUD-PV trial; www.ClinicalTrials.gov identifier #NCT01949805).
For ET patients resistant or no longer candidates for HU or anagrelide, other nonleukemogenic drugs (such as IFN) may be used. Pipobroman is not used as frontline therapy given its leukemogenic potential, as demonstrated in the French Polycythemia Study Group (FPSG) randomized study.37 It may be considered for patients over age 70 who are intolerant to the aforementioned options. Busulfan incurs similar risks and should be similarly restricted to older populations.
In MF, treatment of symptomatic anemia may include the use of corticosteroids, androgens, immunomodulatory agents (thalidomide, lenalidomide), erythropoiesis-stimulating agents, and splenectomy.38,39 It should be mentioned that not all immunomodulatory agents demonstrate similar efficacy. A phase 3 study of pomalidomide versus placebo in transfusion-dependent MF patients showed no significant differences in transfusion responses at 16% (range 11%–23%) versus 16% (range 8%–26%).40 Nontargeted cytoreductive treatments including HU, cladribine, oral melphalan, and busulfan have shown very modest clinical benefits for splenomegaly, but may potentiate leukemic transformation with significant side-effect profiles.41 Nonpharmacological modalities such as radiotherapy or splenectomy are also available for select populations. Given the high mortality rate (5%–10%) and considerable morbidity (47%), splenectomy is reserved for patients with exceptionally large spleens or those failing pharmacological therapies.42
JAK2 inhibition: single agent
Ruxolitinib
Ruxolitinib is approved for MF in the United States, European Union, and many other countries based on the two COMFORT studies in which its effects on splenomegaly, symptom improvement, and perhaps survival were successfully demonstrated41,43 In a 3.5 year follow-up analysis of the COMFORT-II data, ruxolitinib was associated with a 42% reduction in risk of death (hazard ratio = 0.58; 95% confidence interval, 0.36-0.93) compared with best-available therapy (BAT). At 3.5 years, the probability of survival was 54% and 71% in the BAT and ruxolitinib arms, respectively.44 Ruxolitinib has also been shown to promote weight gain (96% of subjects) and to improve total cholesterol (97% of subjects) presumably via reversal of MF-related cachexia and catabolic pathways.45 It is currently under investigation for intermediate-1 disease MF patients in the phase 2 study ROBUST.46
Ruxolitinib has also been evaluated in ET and PV, with most activity seen in patients with PV. In a phase 2 study of 34 PV patients refractory or intolerant to HU receiving ruxolitinib for a median of 152 weeks, hematocrit levels <45% and nonpalpable spleen measurements by 24 weeks were achieved in 97% and 44% of patients, respectively, with significant improvement in symptoms.47 The RESPONSE trial is a randomized phase 3 study evaluating the use of ruxolitinib versus BAT in PV patients resistant or intolerant to HU (www.ClinicalTrials.gov identifier #NCT01243944). By week 32, 71.8% and 23.6% of ruxolitinib-treated patients had a decrease in spleen size and complete hematological remission from baseline compared with 33% and 8.9%, respectively, in the BAT arm.48 The RELIEF study (www.ClinicalTrials.gov identifier #NCT01632904) is an ongoing randomized, double-blind study comparing symptoms in ruxolitinib-treated PV patients versus those receiving HU. Results on this trial have not yet been made publically available.
Other JAK2 inhibitors
Numerous other JAK2 inhibitors exist in various stages of investigation (Figure 3), with 2 currently in phase 3 trials. Momelotinib (CYT387) has demonstrated efficacy in improving splenomegaly and MPN-related symptoms with the added benefit of reducing anemia. A phase 1/2 trial of high- or intermediate-risk MF patients recently demonstrated anemia and spleen response rates of 59% and 48%, respectively.49 Seventy percent of RBC transfusion-dependent patients (1 month before study entry) achieved transfusion independence. However, grade 3/4 thrombocytopenia was noted in 32% of recipients. The drug is currently being evaluated against ruxolitinib in a 24-week double-blind, phase 3 study of primary or secondary intermediate- and high-risk MF patients naive to JAK2 inhibitor therapy (www.ClinicalTrials.gov identifier #NCT01969838).

Pacritinib (SB1518) has proven efficacious in reducing splenomegaly and improving constitutional symptoms, with limited provocation of cytopenias, particularly in patients with baseline thrombocytopenia. In a pooled study of 129 MF patients from the databases of 2 phase 1/2 clinical trials, pacritinib was evaluated specifically for its effects on patients with thrombocytopenia.50 Forty-three percent and 38% of patients with baseline platelet counts <100 000/μL and <50 000/μL, respectively, had a ≥35% reduction in spleen volume from baseline. Consistent with previous data, most adverse events were gastrointestinal related and mild. Forty-six percent of patients with baseline platelet counts ≤100 000/μL showed no significant grade change in hemoglobin and platelet counts from baseline. The drug is currently undergoing comparison with BAT in 2 phase 3 studies of MF patients without (PERSIST-1 [www.ClinicalTrials.gov identifier #NCT01773187]) and with (PERSIST-II [www.ClinicalTrials.gov identifier #NCT02055781]) baseline thrombocytopenia.
Several JAK2 inhibitors have been removed from clinical trials secondary to toxicity (Figure 3). Fedratinib (SAR302503), an inhibitor of JAK2V617F, FLT3, and RET, was removed from clinical trials in November 2013 as a result of neurological complications including Wernicke's encephalopathy. However, its efficacy was substantial in MF and documented in a phase 3 randomized, double-blind placebo-controlled study (JAKARTA).51 Similarly, CEP-701, XL019, LY278544, BMS-911543, and AZD 1480 have been removed from trial secondary to myelosuppression, gastrointestinal toxicity, neurological effects, and/or insufficient activity.
Non-JAK2 targets and combination treatments
Recognizing that MPN disorders are the result of constitutive activation of the JAK-STAT signaling pathway, with subsequent propagation of downstream mediators (STAT3/5, PI3K, MAPK), combination treatments targeting multiple levels of the signaling cascade became a rational intervention. A variety of small-molecule novel therapies have been under parallel investigation with JAK2 inhibitors to emphasize synergistic biological activity and to compensate for treatment-related adverse events such as anemia (Table 2).
Current JAK inhibitor clinical trials for patients with an MPN
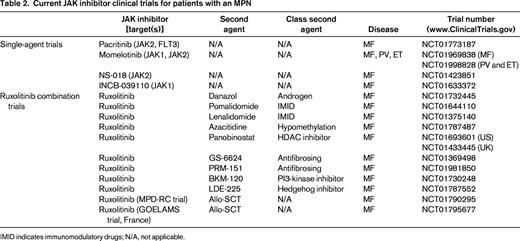
IMID indicates immunomodulatory drugs; N/A, not applicable.
The PI3K pathway is of particular interest given its pleiotropic effects on cellular proliferation, metabolism, and mediation of cellular drug resistance.52,53 PI3K inhibitor use in combination with ruxolitinib has demonstrated significant antiproliferative activity, suggesting that inhibition of JAK2 signaling plays an essential role in increasing sensitization of cells to PI3K inhibition.54 Mammalian target of rapamycin (mTOR) similarly functions as a regulatory serine/threonine kinase important to cellular metabolism, apoptosis, and proliferation.55 Use of mTOR inhibitors in singularity within MPN disorders has demonstrated modest results without affecting JAK2V617F mutational burden, likely reflecting the existence of alternative regulatory pathways.56 Their use as combination treatment with JAK2 inhibitors is under investigation.
Histone deacetylase (HDAC) inhibitors (givinostat) are also a rational MPN therapy considering that HDAC modulates several genes involved in cell cycle regulation, hematopoiesis, proliferation, and apoptosis. An investigation using givinostat showed JAK2V617F+ cells to be 2-3 times more sensitive to givinostat-induced inhibition of colony formation, cellular proliferation, and apoptosis induction than JAK2-wild type cells.57 HDAC inhibitors are currently under clinical investigation with ruxolitinib (www.ClinicalTrials.gov identifier #NCT01693601 in the United States and www.ClinicalTrials.gov identifier #NCT01433445 in the United Kingdom). The hedgehog signaling pathway is involved in cellular differentiation and proliferation in multiple stages of hematopoiesis, along with maintenance and homeostasis of cellular precursors.58 The efficacy of HDAC inhibitors as single agents has been documented in solid tumor malignancies, and their role in MPN disorders remains under investigation (LDE-225; www.ClinicalTrials.gov identifier #NCT01787552). Antifibrotic agents function as endogenous proteins that respond to local tissue damage and stimulate macrophage and monocyte differentiation with subsequent prevention and reversal of fibrosis. In a phase 2 study of MF, the antifibrotic agent pentraxin (PTX-2) showed improvement in BM fibrosis, cytopenias, symptoms, and spleen size.59 Antifibrotic agents are presently being investigated with ruxolitinib in two clinical trials (www.ClinicalTrials.gov identifiers #NCT01369498 and #NCT01981850). Imetelstat, a potent inhibitor of telomerase that uniquely distributes specifically to BM, has been shown to result in molecular remissions and hematological remissions and to reverse BM fibrosis.60,61 Side effects include myelosuppression, with the greatest toxicity observed in patients receiving upfront dosing. Imetelstat is currently on hold for use in ET and PV due to liver toxicity. Immunomodulatory agents have proven efficacious in the treatment of anemia and thrombocytopenia. Their use in combination with ruxolitinib is under active investigation (www.ClinicalTrials.gov identifiers #NCT01644110 and #NCT01375140).
The positive impact of JAK2 inhibitors on splenomegaly, symptoms, performance status, and quality of life makes these therapies a logical adjunct to allo-SCT, particularly as preconditioning. The use of ruxolitinib in allo-HSCT is currently under prospective investigation in 2 clinical trials (www.ClinicalTrials.gov identifiers #NCT01790295 and #NCT01795677).62,63 The JAK ALLO phase 2 study is assessing the impact of ruxolitinib in intermediate- and high-risk MF candidates for hematopoietic SCT.62 The study was briefly halted due to the development of tumor lysis syndrome and adverse events, but has resumed accrual based on interim analysis. A German allo-SCT trial similarly structured to the French study has demonstrated improvements in constitutional symptoms in 86% of patients receiving ruxolitinib at the time of transplantation, with 45% displaying a ≥50% reduction in spleen size.63 At present, JAK2 inhibition before allo-SCT remains experimental and is best done in the setting of a clinical trial such as the MPD-RC 114 trial (www.ClinicalTrials.gov identifier #NCT01790295).
How to treat
PV and ET
As previously discussed, in 2014, the management of PV and ET patients is primarily focused on prevention of thrombohemorrhagic complications and symptom management (Figure 4). In the future, we will perhaps have the molecular insight to choose medical therapies that can definitively prevent the progression of MF or MPN blast phase. At present, we recommend that all PV and ET cases be assessed for prognosis by risk score (Table 1) and MPN symptom burden (MPN-10)21 at the time of diagnosis. In our opinion, stratifying MPN symptom burden by MPN-10 quartile (MPN-10 Q1: <8; MPN-10 Q2: 8-17; MPN-10 Q3: 18-31; MPN-10 Q4: ≥32) simplifies symptomatic groupings and allows for objective evaluation of symptomatic response. Further validation of this approach is ongoing.22 Per ELN recommendations, prevention of vascular events in PV is a crucial goal and is accomplished by controlling hematocrit levels to <45% and using low-dose aspirin.31-33 The role of aspirin in ET is unclear, but may be beneficial in patients with microvascular symptoms. Cardiovascular risk factors should be aggressively managed for both disorders, including smoking cessation if applicable.

Selection of PV or ET patients needing cytoreductive therapy should incorporate both the MPN risk score and clinical scenario (ELN recommendations) such as intolerance of phlebotomy. In our opinion, low-risk patients not receiving cytoreductive therapy should be monitored for changes in symptom burden, vascular events, or pathologic progression, which, if present, might warrant additional therapy. Regular assessment with MPN-10 may be beneficial in determining which patients are symptomatically undermanaged. For both ET and PV, the presence of former or current vascular events automatically constitutes a high-risk disease profile and frontline cytoreductive therapy should be used. In concordance with ELN recommendations, HU is frontline therapy for both disorders with anagrelide or IFN as second-line options in ET and PV, respectively. Clinical trials may also be considered. In our opinion, high-risk patients who, despite receiving standard therapies, develop worsening symptom burdens, persistent vascular events, or cytoreductive resistance or intolerance, should be considered for JAK2 inhibitor or IFN trials with HU (if they have not previously received). Data from the randomized phase 3 trials of ruxolitinib for PV may lead to US Food and Drug Administration (FDA) or European Medicines Agency (EMA) approval and provide a clear second-line therapy for PV patients or for those on HU who have inadequately controlled symptoms.
MF
Management of MF patients requires initial evaluation of the risk score using the choice of IPSS, DIPSS, or DIPSS-Plus (when karyotype available) and symptom burden (MPN-10). We believe that the management of DIPSS low-risk MF patients should be guided by symptomatology for the time being and in the future perhaps by early disease-altering therapy (when shown definitely for either IFN or new agents; Figure 5). It remains unclear whether patients who are considered low risk by DIPSS but harbor high-risk molecular features should be managed differently (ie, earlier choice for allo-SCT). Low-risk patients with low symptom burdens (symptom quartiles 1 and 2) may be observed or considered for a trial with IFN. Otherwise low-risk patients demonstrating higher symptom burdens (symptom quartiles 3 and 4) may be considered for ruxolitinib, JAK2 inhibitor trials, or IFN.

As per ELN recommendations, intermediate- to high-risk patients, regardless of symptomatology, should be urgently assessed for allo-SCT candidacy. Patients deemed candidates may proceed with allo-SCT and, in our opinion, be considered for a JAK2 inhibitor trial first. There is mounting evidence to support this approach in MPN blast phase as well.64 For patients not deemed to be allo-SCT candidates or those in whom allo-SCT may be delayed, it is our opinion that ruxolitinib be initiated or that the patient be considered for enrollment into a JAK2 inhibitor trial. In our experience, using current US labeling recommendations for the ruxolitinib starting dose may lead to anemia or thrombocytopenia in patients with compromised baseline values (ie, hemoglobin <10 g/dL regardless of platelet count). Therefore, we recommend a more modest starting dose of 10 mg twice daily for all patients with a hemoglobin <10 g/dL and platelet counts >100 000 × 109/L and 5 mg twice daily for platelet counts between 50 and 100 × 109/L (ruxolitinib product label). Long-term follow-up of the COMFORT I and II trials do suggest that cytopenias from ruxolitinib use are the worst during the initial 3 months of therapy and that anemia in particular can improve after 6 months. Therefore, it is best to avoid early dose reductions if the patient is stable from an anemia or thrombocytopenia standpoint. If cytopenias become problematic with JAK2 inhibitor therapy (or are not aided by JAK inhibitor therapy), one may consider many ongoing clinical trials that incorporate ruxolitinib plus a second agent to aid anemia (androgens, immunomodulatory drugs, hypomethylating agents, antifibrosing agents). In our opinion, patients nonresponsive to JAK2 inhibitor therapy from a splenic or symptomatic standpoint (including patients with refractory cytopenias) should be considered for trials with other JAK2 inhibitors (in the second-line setting), ruxolitinib combination trials, or clinical trials using non-JAK2 inhibitors.
Conclusion
The past 10 years after the discovery of the JAK2-V617F mutation has led to unprecedented advances in our understanding of MPN biology and many therapeutic innovations. Management of MPN patients has evolved to require a thoughtful and individualized approach that incorporates prognosis, disease burden, candidacy of allo-SCT, assessment of comorbidities, and the patient's therapeutic goals. JAK inhibitors have made a meaningful impact as single-agent therapies for MF and now potentially problematic cases of PV. A broad array of ongoing trials will answer the question of whether alternative pathways (HDAC inhibitors, telomerase, hedgehog) alone or in combination with JAK inhibitors are more effective than JAK inhibition alone.
This article was selected by the Blood and Hematology 2014 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2014. It is reprinted with permission from Blood 2014, Volume 124.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Ruben A. Mesa, Division of Hematology and Oncology, Mayo Clinic, 13400 E. Shea Blvd, Scottsdale, AZ 85259; Phone: (480)301-4502; Fax: (480)301-4675; e-mail: mesa.ruben@mayo.edu.
