
Abstract
MYC, a member of the helix-loop-helix leucine zipper family of nuclear transcription factors, is a potent proto-oncogene primarily identified as the target of the t(8;14)(q24;q32) chromosome translocation in Burkitt lymphoma. Activation of the MYC gene in normal cells both results in enhanced cellular proliferation and up-regulation of pro-apoptotic pathways, reflecting the tight regulation of the molecule in the normal cellular system. In the process of transformation, these secondary inhibitory functions of the MYC molecule have to be overcome through secondary mutations of the MYC gene itself and/or by abrogating the inhibitory effects of physiological regulators and/or repressors of proliferation such as BCL2, BCL6, BLIMP1, or others. Most aggressive lymphomas, therefore, harbor additional oncogenic alterations that cooperate with MYC deregulation, with different alterations identified in human solid or hematological tumors. These alterations are likely to counteract the pro-apoptotic function of MYC. MYC gene alterations in diffuse large B-cell lymphomas and in B-cell lymphomas, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma are frequently associated with BCL2 or/and BCL6 translocations conferring a very aggressive behavior. This review summarizes inherent factors of the biology and function of MYC important in the process of transformation, especially taking account the interdependence of MYC on various cellular networks that have to be co-deregulated to achieve the full malignant phenotype.
Learning Objective
To have insight into MYC structure and function and the mechanisms of its deregulation and the necessary cooperating pathways
Introduction
The expression of the proto-oncogene MYC is deregulated in a large variety of cancers and, in these tumors, overexpression of MYC is often associated with a poor prognosis. The MYC gene is located in band 8q24 in the subterminal portion of the long arm of chromosome 8. It represents a member of the helix-loop-helix leucine zipper family of nuclear transcription factors; other members of this family include MYCN, MYCL, S-MYC, and MYCB. Genes of this family have important functions related to cell growth, survival, and biosynthesis. MYC was identified as the first nonmutated gene that could be activated via retroviral promoter insertion. Subsequently, this finding was the starting point for the identification of other genes that could be activated without occurrence of gene mutations.
In the human system, MYC can be activated via at least 3 modes: insertional mutagenesis, chromosomal translocation, and amplification.1 This review focuses on the impact that MYC has on the proliferative capacity, survival, and, finally, transformation of cells especially taking into account the cross-talk of MYC with other genetic factors cooperating with MYC in the process of transformation, enhancing the transforming potential of the gene.
Normal structure and regulation of MYC
The MYC gene consists of 3 exons. Exon 1 is noncoding and exons 2 and 3 represent the coding regions of the gene. Activation of MYC requires binding the transcription factor MAX, forming a heterodimer that binds to specific DNA sequences in the promoter regions of target genes.1-3 Upon activation, MYC is rapidly transcribed; conversely, the half-lives of both the MYC mRNA and protein are short, ensuring a tight regulation at both the transcriptional and protein level in normal cells. In addition, microRNAs (miRs) have been found to assist in controlling its expression. MYC itself is activated by binding the histone acetyltransferases CBP/p300 and TIP60/GCN5 or the transcription factor P-TEFb, among others. Transcriptional repression of MYC is mediated by interaction with the transcription factor MIZ-1, which prevents recruitment of the activating molecule p300 and enables binding of the gene-silencing DNA-methyltransferase DNMT3a. Other transcription factors such as MAD titrate out MYC from the complexes, followed by the recruitment of histone deacetylases (HDACs) by the MAX/MAD heterodimers that ultimately repress gene transcription.3
MYC represents a global transcription factor that is thought to regulate 10%–15% of genes in the human genome. Therefore, its activation results in what has been termed an “avalanche” effect on a significant proportion of human genes. More specifically, the MYC gene is now regarded as a global amplifier of the transcriptional response (detailed later).4,8,9 MYC controls many aspects relevant to the survival and division of cells, such as DNA replication, protein synthesis, and the regulation of metabolism and energy (Table 1). Activation of MYC results in the direct or indirect activation of the expression of genes that govern the cell cycle and repress cell cycle inhibitors, thus enabling the transition of cells from the G0/G1 to the S phase.1-3 MYC also regulates the expression of miRs that are involved in cell growth and division.5,6 In contrast to these functions, there is also a profound role of MYC in the process of apoptosis.1,3 In the activation of the p53 pathway, MYC/Forkhead box protein (FOXO) complexes activate CDKNA2/p14ARF, thus stabilizing p53 via inhibition of MDM2-mediated degradation.1 In addition, MYC overexpression also results in the induction of the pro-apoptotic molecule BIM. BIM in turn acts via BAX and BAK activation to induce intrinsic mitochondrial apoptosis.1,2 The latter 2 principal pathways are required to counteract the effect that MYC has on normal cells in inducing proliferation and therefore constitute a safeguard against the outgrowth of mutated cells and thus transformation.
Target genes of MYC associated with cellular growth and transformation
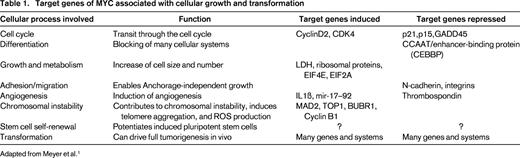
Adapted from Meyer et al.1
It is quite clear that tumor cells with activated MYC must have developed strategies to antagonize these important secondary features of the Janus-headed MYC molecule, thus implying the need for other cooperative mechanisms for cell transformation and tumor progression. In fact, it turned out that loss of function of one of the dual apoptosis pathways is sufficient to abolish full function of the stress response pathways, thus precluding the need for any inactivating factors in the other.7
Cellular functions of MYC
Generally, MYC is believed to function as an amplifier of an already existing gene transcription program in a cell by binding to the promoter regions of actively transcribed genes, rather than activating silenced genes.8,9 The preexisting transcriptional program involved in the functional role of a cell, therefore, will be enhanced by MYC, but not fundamentally altered. This feature may explain the enhanced aggressiveness of lymphoid tumors already associated with a particular gene alteration in which an additional MYC alteration develops. MYC has been shown to function both as an activator and as a repressor of gene transcription. The formation of MYC/MAX heterodimers is the crucial event in this scenario,10 activating gene transcription by several mechanisms. MYC/MAX complexes can bind directly to gene promoters, recruit HDAC complexes to chromatin, alter the chromatin structure through interaction with other proteins, and recruit DNA polymerase II.1 Conversely, the concept of MYC as a transcriptional repressor emerged with the discovery that activated MYC was able to silence the expression of its nontranslocated allele in Burkitt lymphoma (BL). Now, it is clear that MYC represses about as many target genes as it activates, mainly by the interaction of MYC/MAX complexes with transcriptional activators bound to DNA through enhancer or initiator elements displacing coactivators and recruit corepressors. Through these mechanisms, MYC orchestrates a plethora of genes associated with the regulation of the cell cycle and cellular metabolism. MYC has been shown to drive cells from the G0/G1 to the S phase of the cell cycle and to shorten G1 by abrogating the transcription of cell cycle checkpoint genes such as GADD45 and GADD153 and inhibiting the function of cyclin-dependent kinases (CDKs). MYC also promotes cell cycle progression by activation of various cyclins, CDK4, CDC25A, E2F1, and E2F2.1 Examples of target genes of MYC important in the (de-)regulation of differentiation, cell growth and metabolism, adhesion and migration, angiogenesis, chromosomal instability, stem cell self-renewal, and, finally, in transformation, are given in Table 1.
Consequences of MYC activation: lessons learned from BL
MYC is expressed at a low level only in some, not all, proliferating normal cells, and some highly proliferating human cells, like those in the dark zone of the germinal center, do not express MYC at all.11 Therefore, transcriptional activation of the molecule will immediately result in the up-regulation of counteracting regulatory mechanisms to prevent alterations of the cellular homeostasis. Because the transforming power of MYC is intimately associated with continuous deregulation of its expression, these regulatory events, critical both to the MYC molecule itself and to the signaling network to which MYC is addicted, have to be circumvented in the formation of a cancer cell. Continuous activation of MYC in BL, an aggressive B-cell neoplasm, is the classical example of the transcriptional deregulation of a nonmutated gene by bringing its critical regulatory sequences under the control of an nonphysiological promoter. Cytogenetic and molecular genetic studies revealed the occurrence of 8q24 rearrangements in >90% of BL, rendering MYC under the control of the immunoglobulin intron enhancer Eμ. Although the exact number is difficult to indicate because of the occurrence of some particular MYC gene rearrangements (such as insertions), there are some BL cases that are virtually negative for 8q24 rearrangements, in which other mechanisms such as amplifications and/or miR alterations may represent similar mechanisms for the deregulation of MYC.1 Protein overexpression of MYC is a consistent finding in MYC-rearranged lymphoma cells.12
Corroborating earlier findings, a recent study using engineered mice has shown elegantly that, in an experimental setting, the sole transcriptional deregulation of MYC may not be sufficient to induce the full malignant phenotype, most probably because of the dichotomous role of the gene in the induction of proliferation and apoptosis.7 Consistent with this finding, although described to be associated with only few additional cytogenetically visible alterations (“simple karyotype”), individual BL tumors have been found to harbor ∼17 gene mutations per case if examined with sensitive (next-generation) sequencing techniques.13-15 Among those, mutations within the MYC gene itself have been encountered in ∼60% of cases, targeting amino-terminal transactivation domains that either promote the transforming capacity of the gene or increase the stability of the protein via reduced ubiquitin-mediated proteolysis. Given the fact that MYC has been shown to represent a member of a large number of network proteins orchestrating the gene expression program of a cell, it is not astonishing that MYC cooperates with many genes and genetic networks in the process of transformation. As mentioned previously, activation of MYC results in increased proliferation that in turn initiates increased apoptosis via activation of stress-response mechanisms.
Insights into how oncogene-induced stress responses are circumvented by BL tumor cells have largely been gained from mouse models. MYC executes its apoptotic function predominantly through the p53 and BIM pathways.16,17 In the p53 pathway, p53 itself is a frequent target for mutations in BL and in non-BLs characterized by a MYC translocation. In cases with wild-type p53, up-regulation of MDM2 or p14ARF loss has been identified, also leading to impaired p53 function as a tumor suppressor protein. The second fundamental apoptosis pathway frequently targeted in BL, the induction of BIM, cooperates with proapoptotic molecules such as BAX and BAK (Figure 1). In experimental systems, loss of one BIM allele has accelerated the emergence of tumors in MYC-overexpressing mice. Impairment of the induction of the proapoptotic element BIM in BL has been observed via direct and indirect mechanisms.1

MYC-associated pathways in the regulation of proliferation and survival. MYC forms a heterodimer with MAX that activates pathways associated with both cell proliferation and growth and oncogenic stress response. Proliferation is initiated via activation of cell-cycle-associated molecules such as cyclins and cyclin-dependent kinases, among others, and the induction of apoptosis is triggered via up-regulation of BIM and p53. The PI3K-signaling pathway cooperates with MYC in cell proliferation and survival.
MYC-associated pathways in the regulation of proliferation and survival. MYC forms a heterodimer with MAX that activates pathways associated with both cell proliferation and growth and oncogenic stress response. Proliferation is initiated via activation of cell-cycle-associated molecules such as cyclins and cyclin-dependent kinases, among others, and the induction of apoptosis is triggered via up-regulation of BIM and p53. The PI3K-signaling pathway cooperates with MYC in cell proliferation and survival.
Because activation of the NF-κB pathway does not seem to be of major importance in Burkitt lymphomagenesis,4,17,18 the recent identification of the PI3K signaling pathway as a major contributor in MYC-induced lymphomagenesis and cell survival is of pivotal interest. In a new mouse model, the dual constitutive activation of both MYC and PI3K resulted in the generation of lymphomas that phenotypically recapitulated human BL by showing monomorphic medium-sized blastic cells, formation of a “starry sky” macrophage picture, and expressing CD10 and MYC and being negative for BCL2. In addition, the tumors showed an overall gene expression profile that was very similar to human BL.7 Interestingly, tumors generated from this model also recapitulated secondary genetic features intimately connected with human BL, such as a simple karyotype and the acquisition of CyclinD3 mutations recently identified in human BL.13 Given the importance of PI3K signaling in BL, it is of considerable interest that sequencing studies have recently identified novel somatic mutations in BL associated with the activation of the PI3K pathway.13-15 These mutations were detected in ∼70% of sporadic and HIV-associated BL. The most frequent findings were activating mutations in the TCF3 gene in 11% of the cases and inactivating mutations in its inhibitor ID3 in 38%–68% of tumors. The mutational inactivation of ID3 is likely to impede TCF3 in its inhibitory effect, resulting in a net constitutive activation of this pathway. TCF3 and ID3 are both expressed in cells of the dark zone of the germinal center, but not in the light zone, implying that BL cells carrying these aberrations may be retained in this highly proliferating GC compartment,19 preventing differentiation. Because PI3K signaling provides a “tonic” prosurvival signal to B cells downstream of the BCR, genetic alterations stabilizing this signaling pathway are important for the survival of BL cells and, indeed, gene-silencing experiments revealed that these mutations are critical for the survival of tumors and therefore constitute a necessary cooperating mechanism of MYC in the pathogenesis of BL (Figure 2).13

Cooperating gene alterations in MYC-driven lymphomagenesis. MYC is activated by gene translocations or amplifications. Activation of the TCF3/ID3 pathway cooperates with MYC in BL, whereas deregulation of BCL2 is a frequent cooperating mechanism in DLBCL. In ALK+ large B-cell lymphomas, MYC is indirectly up-regulated by the oncogenic effect of ALK and STAT3 activations. As can be seen from the figure, numerous cooperating pathways stabilize MYC, activate coactivators, or deregulate suppressor genes.
Cooperating gene alterations in MYC-driven lymphomagenesis. MYC is activated by gene translocations or amplifications. Activation of the TCF3/ID3 pathway cooperates with MYC in BL, whereas deregulation of BCL2 is a frequent cooperating mechanism in DLBCL. In ALK+ large B-cell lymphomas, MYC is indirectly up-regulated by the oncogenic effect of ALK and STAT3 activations. As can be seen from the figure, numerous cooperating pathways stabilize MYC, activate coactivators, or deregulate suppressor genes.
Aside from its effects of reprogramming the global gene expression profile of transformed cells, MYC is also implicated in the regulation of miRs.20-22 MYC is known to up-regulate an oncogenic group of miRs known as the miR-17-92-cluster. The miR-17-92 polycistron at 13q31 is amplified in many types of lymphoid tumors and exerts its oncogenic function via down-regulation of regulatory genes such as PTEN, TP53, and E2F1.3,20 The consistent regulatory effect on a whole array of miRs exerted by MYC, however, is mainly based on repressing their function; these included miR-22, miR-26a, miR-29c, miR-30e, miR-146a, and miR-150, among others. In this process, MYC frequently associates with promoters of pre-miRNAs and represses miR expression by the recruitment of HDAC,21,22 thereby potentiating the loss and mutation of miR sites found in tumor cells in general. As an example, miR-15a and miR-16-1 are recurrently deleted in lymphoid tumors and target the antiapoptotic molecule BCL2, the expression of which is characteristically down-regulated in BL.
MYC in lymphomas other than BL
Translocations involving MYC or MYC deregulations without translocation are detected also in non-BL tumors, among them follicular lymphoma, diffuse large B-cell lymphoma (DLBCL), and B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL (BCLU). Approximately 5%–15% of DLBCL cases and 30%–50% of BCLU cases carry MYC translocations.23 The number of gene amplifications may be low, with a recent study identifying 2% of DLBCL with MYC amplifications.24 Among DLBCL, 50% of plasmablastic lymphomas, an aggressive lymphoid neoplasm with the phenotype of a terminally differentiated B cells,25 harbor MYC translocations that may be necessary to counteract the repressing effect of BLIMP1 exerted on the proliferation in cells with plasmacytoid differentiation.26 Interestingly, anaplastic lymphoma kinase (ALK)-positive large B-cell lymphomas, aggressive tumors characterized by ALK rearrangements and consecutive overexpression of the ALK protein, express high levels of MYC protein in the absence of MYC gene translocations,27 possibly mediated by STAT3 activation that represents a downstream effector of ALK activated by phosphorylization in ALK+ DLBCL. Similar to what is believed to be the situation in plasmablastic lymphoma, the activation of MYC by STAT3 may be a mechanism to overcome the repressing effects of BLIMP1 on cellular proliferation.
In contrast to the situation in BL, in which MYC is translocated to IG heavy and light chain loci, MYC is frequently rearranged with non-IG genes such as BCL6, BCL11A, PAX5, and ICAROS49 in DLBCL and BCLU, and most MYC-rearranged DLBCL and BCLU cases harbor complex karyotypic alterations.28,29 In addition, ∼60%-80% of MYC rearrangements in DLBCL are accompanied by either BCL2 or BCL6 rearrangements.30,31 The fact that ID3 and TCF3 mutations have not or have only infrequently been identified in the vast majority of DLBCL cases13-15 suggests that these mutations do represent a necessary cooperating mechanism of MYC to create the distinct morphological and biological profile of BL (Table 2).

MYC protein expression is seen in the majority of DLBCL cases, but the number of positive cells varies. MYC protein is highly expressed (80% of cells) in the nuclei of DLBCL with MYC rearrangements12 or amplification; however, only one-third of DLBCL cases with substantial (30%–40% of cells) MYC protein expression carry MYC gene alterations.24,31,39 This suggests that mechanisms other than gene rearrangements are responsible for elevated protein expression in a considerable proportion of DLBCL cases. The mechanism for the transcriptional up-regulation of MYC in translocation-negative and amplification-negative tumors is not clear, but certain miR alterations have been documented in some cases.32,33 The sole protein overexpression of MYC has been associated with inferior prognosis in some studies,24 but, as is the case with MYC translocations, MYC overexpression in DLBCL may not be predictive of an inferior prognosis on its own because there is good evidence that it is the dual deregulation of both MYC and BCL2 expression that is strongly correlated with shorter survival.24,39,40 Immunohistochemical expression scores using MYC and BCL2 have also been found to identify patients with poor prognosis within International Prognostic Index subgroups.24,31,39
BCLU is primarily defined by morphological features. However, MYC rearrangements can be detected in 30%–50% of these tumors,23,34 and this provisional category in the World Health Organization classification harbors the largest number of cases characterized by dual or triple translocations involving MYC, BCL2 or BCL6, or both. On purely morphological grounds, these tumors lie on the crossroads between BL and DLBCL, and gene expression profiling analyses have also shown a profile intermediate between BL and DLBCL in some of the “double-hit” (DH) or “triple-hit” lymphomas; others display a BL gene expression profile.4 Interestingly, a recent publication identified ID3 mutations in MYC/BCL2 or MYC/BCL6 DH lymphomas of both BCLU and DLBCL morphology,35 so these tumors may indeed be viewed as an intermediate category of lymphoid tumors on the genetic level as well, displaying genetic alterations encountered both in BL and DLBCL. As is the case with MYC-rearranged DLBCL, our knowledge on the nature, sequence, and consequences of genetic events in this still incompletely understood category of aggressive lymphomas is still sparse. Because MYC/BCL2 DH lymphomas can also arise as secondary tumors transforming from low-grade follicular lymphomas, we can anticipate that the differentiating effect of the BCL2 translocation on germinal center B cells leading to a marked down-regulation of the proliferation in the neoplastic follicular cells is abrogated by the secondary deregulation of the MYC gene locus. Therefore, the concomitant deregulation of both oncogenes is an important genetic pathway in the emergence of this aggressive neoplasm, which represents a tumor category largely refractory to established therapy protocols.36,37 Most DH lymphomas harbor concomitant MYC and BCL2 gene translocations, but a minority of them have MYC/BCL6 rearrangements, and these tumors have distinct features compared with their BCL2-rearranged counterparts.38 The cooperating effect of the BCL2 protein with MYC to provide prosurvival signals is also mirrored in the fact that it is the dual expression deregulation of both genes that has strongly been correlated with shorter survival in different study cohorts of DLBCL,31,39-41 thus making it a robust prognostic marker in the absence of chromosomal translocations.
Therapeutic options
As has been found recently, the function of MYC depends on the regulatory function of BRD4, a member of the bromodomain and extraterminal (BET) subfamily of proteins that recruit elements required for transcription. The administration of 2 small molecules, JQ1 and iBET, that displace BRD4 from acetylated chromatin have been shown to result in a down-regulation of MYC transcription and in a modulation of its transcriptional program, resulting in an antiproliferative cell effect and tumor growth inhibition in several MYC-addicted hematological tumors such as plasma cell myeloma and BL cell lines with translocated IGH/MYC and also in aggressive lymphomas with MYC overexpression not related to structural gene alterations, suggesting that this might be an exploitable therapeutic strategy in a broad spectrum of MYC-driven tumors.42-44
Summary
Transcriptional deregulation of MYC is the biological hallmark of BL and, in this disease, it is normally associated with few secondary chromosomal alterations and characteristic somatic mutations stabilizing MYC, activating cell-cycle-associated factors such as CCND3, and coactivating the PI3K pathway via TCF3 and ID3, among others. The molecular consequence of these characteristic cooperating features is to counteract the inherent pro-apoptotic functions of the MYC molecule. In contrast, in non-BLs, the transcriptional deregulation of MYC represents a secondary event associated with complex karyotypes and a large mutational load in tumors such as DLBCL and BCLU, and these are frequently accompanied by BCL2 or BCL6 translocations. Judging from the still rudimentary clinical data, the dual or triple deregulation of these oncogenes results in the formation of highly aggressive tumors that are often resistant to current therapy protocols. A substantial number of DLBCL cases, and possibly also other aggressive lymphomas, display MYC protein overexpression not mediated by translocations. The concomitant overexpression of the BCL2 protein is the critical adverse factor associated with inferior survival in these tumors, again stressing the importance of the deregulation of several oncogenic pathways overcoming the inherent blocking mechanisms even in genes with “built-in” safeguard mechanisms. The integration of these new genetic findings, and especially the improved insight into the regulatory network of MYC, may help to overcome the dismal prognosis of these malignancies, opening possibilities to target several critical components of regulatory networks at a time.
Acknowledgments
The author is supported by the Robert Bosch Foundation, Stuttgart, Germany. The author thanks Mrs. Elisabeth Ott for artful graphic work.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
German Ott, Department of Clinical Pathology, Robert-Bosch-Krankenhaus and Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Auerbachstrasse 110, 70376 Stuttgart, Germany; Phone: 49-711-8101-3390; Fax: 49-711-8101-3169; e-mail: german.ott@mail.rbk.de.
