
Abstract
Acute lymphoblastic leukemia in childhood has shown remarkable improvements in outcome over the past decades. This achievement was the result of better patient risk assessment, intensification of treatment, appropriate use of BM transplantation, and improved supportive therapies. Among risk factors, early response (originally morphologic and today minimal residual disease) has acquired a prominent role. The predictive value of minimal residual disease evaluation as a measurement of in vivo drug resistance opened new perspectives for its use in clinical evaluation to determine a risk-based treatment and as a potential surrogate end point for efficacy. More recently, detailed genomic analyses of childhood acute lymphoblastic leukemia have increased our knowledge in this disease. It is likely that this will lead to further improvement of risk assessment and stratification to targeted therapies. Leukemic subsets defined on the basis of biological mechanisms and driver mutations will be ever smaller. To facilitate continued progress, this new scenario will raise methodological issues in study design and the need for collaboration across large, well-characterized patient populations.
Introduction
Over the past 3 decades, remarkable advances have been achieved in the treatment of acute lymphoblastic leukemia (ALL) in children.1,2 In contemporary clinical trials, the 5-year survival rate has risen above 85% in developed countries.1 This achievement is due at least in part to better defined patient risk classification, optimization of antileukemic agents, and improved supportive care.3 Furthermore, the extraordinary impact of new genomics4 and the availability of new targeted treatment modalities are likely to further improve the overall survival rate of children with ALL. Here, we discuss the many lessons learned from the most recent ALL trials, with particular emphasis on the molecular characterization of the early response as a prognostic parameter for risk stratification, which points to an increasingly important role of genomics in ALL research.
Lessons from current clinical ALL trials
The term “risk stratification” is used to define the process of allocating patients to specific treatment modalities on the basis of their probability of presenting with a relapse when treated with conventional “standard therapy.” Therefore, patients with very low risk of relapse are likely to be good candidates for reduced-intensity treatments to prevent early or late severe toxicity, whereas those at high or very high risk of relapse might benefit from intensified or markedly intensified therapies, which could also include allogeneic hematopoietic stem cell transplantation (SCT). Stratification factors can thus be regarded as “relative” prognostic factors because the actual prognosis is eventually determined by what type of treatment is given, which could overcome the impact of the prognostic factors considered for the stratification itself. For this reason, stratification factors have been modified in recent years, leading to improved results, and may still be further optimized depending on the overall treatment intensity, cumulative doses of specific drugs, and the aims of the study protocols in use.
Factors such as age, WBC count at diagnosis, or sex may still be relevant in stratifying patients in protocols that do not include intensive initial therapy, but rather deliver protracted and high cumulative doses of vincristine and glucocorticoids. In addition, the T-cell immunophenotype may still be an important stratification factor when high doses of methotrexate and/or of dexamethasone are not used. Likewise, there may be little utility in identifying very-low-risk patients on the basis of factors such as hyperdiploidy, favorable trisomies, age, WBC count,5,6 and early response to therapy when the initial treatment is rather intensive for all patients.
Another aspect that may be relevant for stratification and treatment is the outcome after relapse. Some subgroups of ALL (eg, T-cell ALL) have very limited chances of rescue after disease recurrence. Conversely, other subgroups (eg, ETV6/RUNX1-positive ALL) are likely to be cured when relapse occurs in patients who did not receive a frontline intensive therapy. In such cases, inferior event-free survival (EFS) may not lead to worse survival, indicating that for some subgroups, survival might be more important than EFS. Therefore, some of the most favorable prognostic factors may only be relevant if the final goal is to reduce treatment burden to a minimum, accepting in some circumstances even a higher risk of relapse. On the contrary, some very unfavorable prognostic factors may become useful only if more intensive treatments are associated with better results or if specific and targeted therapies are or will be available. This is the case, for example, for Philadelphia chromosome-positive (Ph+) ALL. Current study protocols take into account the above-mentioned considerations to achieve the most suitable stratification while tailoring the treatment as much as possible. For this purpose, separate treatment protocols have been developed for ALL subgroups such as Ph+, T-cell, and infant (< 1 year of age at the diagnosis) ALL. Even in these patients, however, it has been clearly demonstrated that the response to induction therapy as determined by minimal residual disease (MRD) level at different time points remains the strongest independent factor to predict both favorable and unfavorable outcome.
The term MRD has been used to define the lowest level of disease detected in patients in continuous complete remission (CR) by conventional methods of analyses (for review, see Campana et al7 ). The use of MRD tests has become prominent in ALL management (for review, see Cazzaniga et al8 ). The main reasons for this development have been the progressive improvement of standardized methodologies applicable to virtually all patients and the conduct of clinical studies that used MRD evaluation as a marker of in vivo early response to allocate patients into different risk-based treatments to improve outcome. Several studies in childhood and adult ALL have identified MRD as the most relevant and independent prognostic factor for the duration of CR.7 There is a close association between the quality of the molecular remission (ie, clearance of leukemia blasts) and the final outcome regardless of the applied treatments. However, it still remains to be determined why the exposure to only a few drugs during the early phases of treatment (induction or consolidation) reveals different in vivo chemosensitivities that influence the final treatment outcome.
In the Italian Association of Pediatric Hematology and Oncology– Berlin-Franfurt-Münster ALL2000 (AIEOP-BFM ALL2000) study, stratification was largely based on PCR-MRD measured at the end of induction (phase IA, time point 1 [TP 1]) and of consolidation (phase IB, TP 2). Patients with negative MRD at both time points were allocated to the standard-risk group, those with an MRD level ≥ 10−3 at TP 2 to the high-risk group, and the other patients to the medium-risk group. Intriguingly, the results obtained in these patient cohorts stratified according to the MRD response cast doubt upon the value of another traditional BFM poor prognostic factor, namely the prednisone poor response, but do not call into question the failure to obtain a CR (morphological) at the end of phase IA.9,10
On the contrary, other factors such as hypodiploidy (< 45 chromosomes), which were not considered in the AIEOP-BFM ALL2000 study, or the presence of the t(4;11) translocation have retained an independent negative prognostic value. In the same study, the MRD level was also monitored by flow cytometry (FCM) at an earlier time point, but it was not used for stratification.11 At day +15 of phase IA, an additional cohort of patients, identified neither by PCR-MRD at TP 2 nor by the presence of hypodiploidy or t(4;11) translocation, showed high levels of residual disease (> 10% BM blasts) and had a poor outcome. Therefore, in the current AIEOP-BFM ALL2009 study, compared with the previous one, high MRD measured by FCM at day +15 and hypodiploidy are being considered as additional factors to identify patients at high risk of relapse and therefore are receiving intensified therapy.
Over the past decade, many different subtypes of childhood ALL have been identified through molecular biology techniques. These subtypes bear several features, such as iAMP21, CRLF2 rearrangements, IKZF1 alteration, JAK1/2 mutations, BCR-ABL1-like signature, and early T-cell precursor (ETP), which are associated with poorer outcome and therefore have been given particular attention.1 However, the predictive value of these features varies among different studies. Furthermore, their independent value with respect to MRD as measured currently in the AIEOP-BFM ALL2009 study (PCR + FCM) remains difficult to be defined considering that also treatment is different from the previous AIEOP-BFM ALL2000 study.12 Therefore, further prospective investigations will be needed to assess whether patients can benefit from treatment intensification or, if available, novel targeted therapies.
Another aspect of risk stratification concerns the indications for SCT. When the results are analyzed taking into account the waiting time for transplantation, patients undergoing SCT in first CR have only a modest advantage in terms of EFS (ranging from 10% to 20%) compared with patients with the same features undergoing chemotherapy.13 Therefore, considering that the sequelae of SCT may be more pronounced than those of chemotherapy and that some HR patients can be rescued after a relapse, indications for transplantation remain controversial. Most recent childhood ALL studies in fact have dropped many of the indications to transplantation used in the 1990s to limit to patients with a poor MRD response eligibility to SCT, which in turn should be performed only after clearing MRD or reducing it to lower levels.
Genomic analysis to drive tailored therapy
The recent availability of genome sequence and adequate analytical platforms, such as gene expression profiling, single nucleotide polymorphism arrays, and, more recently, next generation sequencing, has expanded the full repertoire of genetic lesions in childhood ALL.1,14
Although many of these genomic studies have increased our knowledge of the pathogenesis of the disease significantly,15,16 there is no consensus on how new specific genotypes will affect the clinical management of children with ALL. It is likely that genomics will be translated into a better risk stratification that will drive tailored therapy in the near future. However, 2 major points need still to be addressed: (1) the impact of genomics in predicting response to therapy and thus to refine risk stratification and (2) how the identification of new targets will be translated into effective targeted therapy. Several factors should be considered in determining clinical significance and prognostic significance of a novel genetic discovery: treatment context, independent verification in multiple prospective clinical trials, independent prognostic value in multivariate analysis with MRD, and importance of the novel genetic aberration either as potential targets or as modifiers of specific therapies.14
The standard classification schema of childhood ALL according to immunophenotype, which has been further subdivided for the presence of numerical and large structural chromosomal aberrations, is continuously being refined by detailed profiling of submicroscopic alterations and mutational analyses.15,16 This has allowed the discovery of new ALL-specific entities that lack detectable cytogenetic alterations by conventional methods and others characterized by the coexistence (and cooperation) of multiple genetic lesions with well-known chromosomal alterations (Table 1). Overall, standard and genome-wide analyses can identify primary genetic abnormalities in 75% to 80% of childhood ALL cases1 (Figure 1).
Novel genomic alterations with prognostic or therapeutic relevance
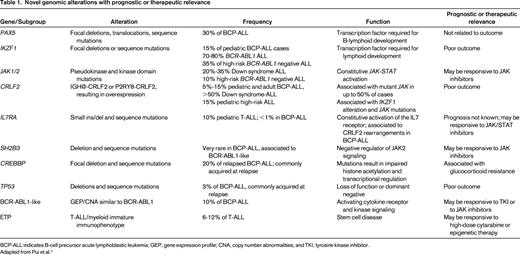
BCP-ALL indicates B-cell precursor acute lymphoblastic leukemia; GEP, gene expression profile; CNA, copy number abnormalities; and TKI, tyrosine kinase inhibitor.
Adapted from Pui et al.1

Estimated frequency of specific genotypes in childhood ALL. In green are the genetic lesions associated with BCP-ALL, in blue are lesions associated with T-ALL. Darker green or blue colors indicate subtypes associated with poor prognosis. The sum of percentages does not take into account the possible coexistence of different lesions. Data have been modified from Pui et al.1
Estimated frequency of specific genotypes in childhood ALL. In green are the genetic lesions associated with BCP-ALL, in blue are lesions associated with T-ALL. Darker green or blue colors indicate subtypes associated with poor prognosis. The sum of percentages does not take into account the possible coexistence of different lesions. Data have been modified from Pui et al.1
Identification of new genomic alterations with potential prognostic relevance
Ikaros (IKZF1) and B-cell development gene deletions
Genome-wide single nucleotide polymorphism array analysis revealed frequent mono-allelic deletions of genes regulating B-cell development, including PAX5, EBF1, and IKZF1. Among them, IKZF1 deletions are frequently associated with the BCR-ABL1 fusion gene (70%-80% of BCR-ABL1–positive ALL),17,18 whereas in BCR-ABL1–negative ALL they occur at lower frequency (10%-15%).19-21 Deletions can affect the whole gene or may involve different exons of one allele, thus creating a dominant-negative isoform.22 The presence of deletions in IKZF1 are associated with a markedly inferior prognosis in childhood17,18 and adult23 BCR-ABL1–positive ALL. Although the IKZF1 alteration itself is an independent prognostic factor of the hazard of relapse in childhood BCR-ABL1–negative ALL, its impact is largely reduced when MRD is considered, making questionable the need and benefit of introducing IKZF1 deletions as additional stratification marker in the context of MRD-based protocols.20,21 Moreover, most IKZF1-deleted cases stratify into the MRD-based high-risk relapse group,21 suggesting that their identification would require an alternative treatment that is still not available. In contrast, Kuiper et al24 have shown that integration of both MRD and IKZF1 can provide a stronger prognostic value than each of the established risk factors alone, allowing prediction of 79% of all the relapses with a 93% specificity. Based on this study, the current Dutch Childhood Oncology Group (DCOG) clinical protocol requires a longer maintenance treatment for MRD-medium-risk patients with IKZF1 deletion. To date, this is the first example of the integrated use of genomics and MRD data (R. Pieters, personal communication). Interestingly, in high-risk relapse patients, deletion of IKZF1 is strongly predictive of a second relapse after SCT and therefore should be considered in future risk assessment.25 None of the other B-cell developmental genes (ie, PAX5, ETV6 or EBF1) are associated with a significantly worse prognosis.
CRLF2-JAK-STAT signaling
Four independent studies (for review, see Izraeli et al14 ) identified aberrant expression of the cytokine receptor CRLF2 and activation of JAK-STAT signaling in approximately 5% to 15% of BCR-ABL1–negative childhood and adult ALL and in approximately 60% of children with Down syndrome ALL. This expression results from either chromosomal translocations of CRLF2 into the IGH locus or from deletions at the pseudo-autosomal region of chromosomes X and Y fusing the coding region of CRLF2 with the first exon of the constitutively expressed P2RY8 gene. CRLF2 is normally associated with the IL7 receptor alpha (IL7Ra) to form the heterodimeric receptor of the inflammatory cytokine TSLP. Aberrant expression of CRLF2 in B-cell precursor (BCP)–ALL is associated with additional somatic events that activate the JAK-STAT pathway. These include activating mutations in JAK1, JAK2, or SH2B3 or activating mutations of either the CRLF2 or IL7RA chains of TSLP receptor itself (for review, see Izraeli et al;14 Palmi et al;12 Roberts et al26 ). These events cause constitutive activation of the JAK-STAT signaling pathway, leading to a growth advantage. Several groups have reported an MRD-independent worse prognosis for patients displaying either CRLF2 overexpression (for review, see Izraeli et al14 ) or CRLF2-P2RY8 fusion.12 Although this subset of BCP-ALL patients, currently stratified as MRD medium risk, could be considered for treatment intensification, their outcome and overall survival did not prompt clinicians to allocate them to the high-risk category. It is highly likely that over the next few years, the relevance of aberrant JAK-STAT signaling for risk stratification will be clarified.
Alterations of the TP53 gene
Copy number and sequence alterations of TP53 were observed in 12.4% of patients with BCP-ALL and 6.4% with T-cell ALL at first relapse, with half of them being gained at relapse.27 In both intermediate-risk and high-risk relapse BCP-ALL patients, TP53 alterations were predictive of poor treatment response, EFS, and overall survival rate. Furthermore, multivariate analysis identified IKZF1 deletion and TP53 alteration as independent predictors of inferior outcome.25 Therefore, IKZF1 and TP53 are most likely to represent relevant prognostic factors to be considered in future risk assessment of children with relapsed ALL. A high frequency of TP53 alterations can occur in both pediatric and adult low-hypodiploid ALL (91.2% and 90.9%, respectively). Although of limited clinical relevance due to the very low incidence, a significant proportion of the TP53 mutations identified in pediatric low-hypodiploid ALL were present as heterozygous mutations in remission BM or peripheral blood and in purified normal T-cell populations, suggesting that in these cases, TP53 mutations are inherited as Li-Fraumeni syndrome and that low hypodiploidy could be a manifestation of this disease.28
Alterations of the CREBBP gene
Sequence or deletion mutations of CREBBP, encoding the histone acetyltransferase CREB-binding protein, were found in 18.3% of BCP-ALL relapse cases.29 These mutations were either present at diagnosis or were acquired at relapse (some being present in subclones at diagnosis) and resulted in truncated alleles or deleterious substitutions in conserved residues of the histone acetyltransferase domain. Functionally, the mutations impaired histone acetylation and transcriptional regulation of CREBBP targets, including glucocorticoid-responsive genes. Therefore, this finding raises the possibility of using epigenetic treatment (eg, DNA methyltransferase and histone deacetylase inhibitors) in these BCP-ALL relapsed patients.
Identification of new BCR-ABL1–like entities with potential therapeutic relevance
A subgroup of approximately 15% of BCP-ALL with a gene expression signature highly similar to the BCR-ABL-ALL (“BCR-ABL1 like”) was reported by 2 independent studies.19,30 This subgroup was associated with a significantly inferior prognosis in independent clinical protocols. IKZF1 and CRLF2 aberrancies (with or without JAK mutations) were present in most of these BCR-ABL1–like leukemias; however, the precise definition of the driving event(s) of this subgroup is still not known. Whether it may actually reflect the fingerprint of the aberrancies of IKZF1 and CRLF2 genes or if it arises from the activation of other kinases acting in a similar fashion as BCR-ABL1 is currently under investigation. Among genetic abnormalities identified in BCR-ABL1–like cases, EBF1-PDGFRB or NUP214-ABL1 fusion responded to ABL1 tyrosine kinase inhibitors (which also inhibit PDGFRB) and BCR-JAK2 or mutated IL7R responded to JAK2 inhibitor in preclinical studies.26
ETP-ALL
A stem cell-like gene expression signature was identified in 12% of T-ALL cases treated in 3 consecutive clinical trials at St. Jude's Hospital and validated in an AIEOP series.31 This signature was associated with a specific immunophenotype characterized by a lack of CD1a and CD8 expression and weak CD5 expression, with expression of stem cell and/or myeloid markers. The risk of relapse in this subgroup was 72% compared with 19% of the other T-ALL patients. Because ETP T-ALL can be easily diagnosed by FCM, it is likely to be translated into clinical practice.
Host pharmacogenomics
Ongoing pharmacogenomics studies hold great promise to yield genetic polymorphisms that could be used to individualize the dosages of antileukemic agents. So far, the only well-established clinical effect refers to mercaptopurine and the genetic polymorphism status of the thiopurine methyltransferase (TPMT) gene.32 Indeed, tailoring the dosages of methotrexate and mercaptopurine to the limits of tolerance has been associated with a better outcome.33 Therefore, customizing the dosage of mercaptopurine based on preemptive testing for thiopurine methyltransferase status will likely decrease the risk of mercaptopurine-induced toxicities associated with an inherited TPMT deficiency. This, in turn, might reduce the likelihood of acute myelosuppression (without compromising disease control) and the risk for the development of mercaptopurine-induced myeloid malignancy.32,34 Moreover, thiopurine methyltransferase also has a significant impact on the pharmacokinetics of thioguanine, because patients with this enzyme deficiency are at an increased risk of developing hepatic venoocclusive disease. It is possible that, in the near future, genetic guidance might ensure a better usage of the standard drugs, improved efficacy, and reduction of toxicity and long-term side effects.35
Implications for future clinical trial design
The increasing number of genomic alterations that have been discovered recently raises 2 important issues regarding the design of future studies. The first is whether these genotypes can be used to improve patient stratification. For example, in a frontline protocol stratified into 3 groups (low, intermediate, and high risk), adding a new stratification factor would be worthwhile only if this increased the separation between risk groups while decreasing the within-group heterogeneity in terms of outcome. Therefore, the addition of a new factor should be done only upon careful evaluation of the following points: (1) the distribution of the factor in the patient population; (2) the significance of its role in terms of prognosis in combination with other consolidated criteria (eg, MRD, WBC counts, etc); and (3) the likelihood that changing risk group (and thus the treatment intensity) for some patients may improve their outcome. This evaluation should also be performed to avoid unnecessary complexity in the stratification system, which is relevant for trial design, feasibility, and generalizability of the results.
The second issue concerns the role of new discoveries in genetics and molecular biology in determining specific treatment modalities. If a specific type of genetic alteration is found to be related to distinct sensitivity of the patient to a targeted therapy, then it could provide the rationale for the creation of a new separate subgroup of patients. This would then lead to a redefinition of the specific treatment protocol for this subgroup regardless of how rare it might be.
Refining treatment stratification and defining subgroups for targeted treatments are just the tip of the iceberg of a more complex reality we have to face in study design. We know that patient stratification is also influenced by available treatment options and that the matching between target therapies and genetic profile of individual tumors is not deterministic. Altogether, the progress in genetics and biology (and biotechnology) that drives the “personalized” treatment approach makes it more and more difficult to gain sufficient evidence regarding new treatments unless these are markedly superior to those used routinely. With this latter exception, when dealing with treatment decisions in rare subgroups, it is often difficult to have sufficient numbers and adequate power for robust conclusions. An example is the subpopulation of children with ALL who, after the induction and consolidation phases, still show persistence of the disease either at the molecular or morphological level. This high-risk subpopulation is small and has a dismal prognosis after transplantation. For all of these reasons, this subpopulation represents a challenging target for investigators, who need an adequate sample size to provide evidence on treatment effect; however, there is room for marked improvements and testing of new promising drugs. For example, these patients may be eligible for phase 2 or 3 studies, which would include novel drugs that have already gone through early phases of clinical development in adults. An example of a rare subpopulation specifically identified in relationship with a targeted therapy is that of Ph+ ALL patients, accounting for only 3% of the ALL population. These patients are now routinely exposed to a tyrosine kinase inhibitor on top of chemotherapy because phase 2 and 3 studies have shown an improvement over the historical 50% 4-year EFS. However, the sample size needed in the framework of a traditional study is difficult to achieve in a reasonable time frame considering that high-risk leukemias often represent a small subset and that, in general, lymphoblastic leukemia is not regarded as a common cancer. For this reason, mainly in childhood leukemia, many countries have developed national study groups that design and run multicenter clinical trials. In addition to that, they have developed international collaborations to address therapeutic questions in trials for rare subgroups, including Ph+ ALL.36 This has allowed researchers to conduct a randomized trial in such a rare setting and has led to the creation of a network that will cooperate again in future studies. From a methodological point of view, whenever possible, it is important to run international trials to reach a high level of evidence that is sufficient for driving changes in treatment practice. Furthermore, it should be taken into account that relatively large trials are also needed to optimize treatment modalities by aiming at modest differences that nonetheless are clinically important for outcome improvement (efficacy), together with a more efficient definition of therapeutic strategies that use existing chemotherapeutic agents.
These studies are generally powered to detect less than 10% absolute increase (or difference) in EFS in a subpopulation of patients, usually of relevant size, who have a relatively good prognosis. In childhood ALL, these studies typically address the so-called intermediate-risk patients, accounting for approximately 50% of the ALL population, who have a 4-year EFS of 70% to 80%. These studies may ask a randomized question on intensification or inclusion of new formulations of existing drugs such as, for example, pegylated L-asparaginase instead of the native product. Treatment optimization may also be achieved by testing strategies that are at least as efficacious as those currently used, but carry a lower burden of toxicity and complications. Typically, this generates studies of noninferiority targeted to subpopulations of patients with good clinical outcome. For example, in childhood ALL, these studies would target patients with an approximately 90% 4-year EFS, for whom the attempt is to de-intensify certain therapy elements with known short- or long-term side effects without compromising the EFS outcome. In addition, in these 2 settings, international cooperation is needed in addition to innovative approaches of study design and conduct.37,38
The definition of the full genetic repertoire of ALL and the progressive availability of new targeted therapy will make the subgroups of patients smaller and smaller. With that in mind, we should always consider the possibility of conducting international collaborative traditional trials to maximize recruitment, even if this would cause a significant increase in the organizational and regulatory burden. To this end, alternative statistical approaches may be needed. It is also essential to give more emphasis on the estimation of treatment effect rather than on the hypothesis testing so that important information can be derived with interval estimates despite a measure of uncertainty inherent in relatively small studies. The use of a Bayesian approach to the study design has also been proposed, because this enables information gathered from previous studies to contribute to the estimation process. This could further reduce the uncertainty, but the validity of this approach relies heavily on the accuracy of the prior information.39
Research institutions and medicine regulatory agencies have issued guidelines on how to run clinical trials in small populations for drug development.40 Although they acknowledge that there are no special methods for the design and analysis of these trials, they agree to the use of less conventional and/or less commonly known methodological approaches if they are useful to improve the study. In the regulatory approval process, deviations from standard randomized controlled trials should only be considered when completely unavoidable and justified.
A final consideration is concerning the clinical use of MRD assessment. Although we can assume that MRD provides relevant additional information on the activity of new drugs because it measures remission in a more sensitive manner, it must be stressed that more research is needed to assess the potential of MRD to replace long-term EFS or survival estimates in pediatric leukemias, (ie, to use MRD as a surrogate marker for “efficacy” assessed as overall survival/EFS). Formal validation of a surrogate end point is usually carried out by performing a meta-analysis of all of the randomized trials in which a new drug (or a class of drugs) was studied, with the aim of assessing whether the treatment effects on MRD are strongly related to the treatment effects on the clinical benefit end point. This would be necessary before using MRD as a primary end point (as a surrogate of survival) in clinical trials.
Conclusions
The remarkable advances in the treatment of childhood ALL have become a paradigm of success in modern oncology. However, we still have to deal with a treatment failure rate of 10%-15% and the need to minimize long-term health complications in a large population of leukemia survivors. To face these challenges and to keep making progress against childhood ALL, we need new drugs and transnational collaborations across large and well-characterized patient populations.
Acknowledgments
This work was supported by the Associazione Italiana Ricerca sul Cancro (IG-8666 to A.B., IG-13574 to G.C., and a 5x1000 grant to A.B.), the Fondazione Cariplo, the Italian Ministry of Health, and the Italian Ministry of University and Research. We thank Maria Grazia Valsecchi and Valentino Conter for conceiving, writing, and revising the manuscript.
Disclosures
Conflict-of-interest disclosure: A.B. declares no competing financial interests. G.C. is employed by Fondazione M. Tettamanti ONLUS. Off-label drug use: None disclosed.
Correspondence
Andrea Biondi, Department of Pediatrics and Centro Ricerca Tettamanti, University of Milano-Bicocca, S. Gerardo Hospital, Fondazione MBBM, 20900, Monza, Italy; Phone: 0039-039-2333513; Fax: +39-039-2301646; e-mail: abiondi.unimib@gmail.com.
