
Abstract
It is well known that signals emanating from the B-cell receptor (BCR) activate downstream pathways to regulate the development and survival of normal B cells. In B-cell malignancies, it is increasingly understood that similar pathways are activated through both tonic and chronic active BCR signaling to promote tumor viability and resistance to therapy. Recently, several active and oral agents have emerged that target key proximal kinases in the BCR pathway, including Bruton tyrosine kinase, PI3K, and spleen tyrosine kinase. In early clinical studies, these agents have shown significant activity across a broad range of B-cell lymphomas and chronic lymphocytic leukemia. Especially impressive responses have been reported in mantle cell lymphoma and chronic lymphocytic leukemia, and many patients remain on treatment with continued disease control. Toxicity profiles have been mild in the majority of early studies, without significant myelosuppression over prolonged dosing. Due to these attractive attributes, several agents targeting the BCR pathway are now entering early combination studies with traditional chemotherapeutics and/or other novel agents. It is clear that agents targeting the BCR pathway will significantly affect the design of future therapeutic regimens for B-cell malignancies. Future research will focus on understanding potential mechanisms of resistance, identifying biomarkers of response, and defining optimal combination regimens.
Introduction
B-cell non-Hodgkin lymphomas (NHLs) and leukemias comprise a complex group of malignancies with various clinical, histopathologic, and molecular features, as well as heterogeneous outcomes after standard therapy. Patients who require treatment often receive combination regimens with genotoxic agents and/or immunotherapeutics. Although initially effective in most cases, this approach is often complicated by significant short- and long-term toxicities including end-organ damage, myelosuppression, and secondary cancers. Relapse or transformation of indolent disease is not uncommon. Salvage therapy is often associated with progressive resistance, and B-cell lymphoma/leukemia remains one of the leading causes of cancer death in the United States.
Therapies that target key cellular pathways/attributes specific for tumor cells are envisioned as a better way to treat cancer. In certain diseases, such as chronic myelogenous leukemia, targeted therapies have substantiated this vision. It has long been suspected that the B-cell receptor (BCR), the defining attribute of normal and neoplastic B cells, would be an effective target in BCR-expressing malignancies. Recent years have seen a convergence of new preclinical evidence that BCR signaling is critical to most B-cell malignancies, the development of clinic-ready targeted agents inhibiting BCR-activated signaling pathways, and clinical trials demonstrating the striking effectiveness of these agents. Despite the success of BCR-targeting therapy for B-cell malignancies, summarized in a recent review article,1 questions remain about how best to translate BCR-targeting therapy to the clinical setting. This review discusses briefly the molecular biology of BCR signaling in B-cell malignancies, presents an update of the clinical experience to date with BCR-targeting drugs, and addresses some questions concerning their further clinical development.
Molecular biology of BCR signaling in B-cell malignancies
A B cell is defined and created by the productive rearrangement of immunoglobulin heavy (IgH) and light (IgL) chain genes, leading to expression of a BCR. The sequence of its IgH and IgL hypervariable regions (HVRs), which determine the specificity and affinity of the BCR to bind to antigenic determinants, is the unique molecular fingerprint of each B cell and its clonal relatives. Signals emanating from the BCR act through downstream signaling pathways to direct the developmental stage, expansion, and survival of a normal B cell. These pathways are also frequently used by B-cell malignancies to drive proliferation, growth, and survival and are the targets of current BCR-related therapeutic approaches, rather than the BCR itself or its generation of signals.
“Active” BCR signaling, in which the BCR is activated by binding of antigen recognized by one or both of the combined IgH and IgL HVRs, is the best understood form of normal BCR signaling. Because the cognate antigen for a particular BCR is usually not known, this binding is often experimentally modeled with an antibody reagent derived from immunoglobulin generated in a nonhuman species and directed against constant regions of human IgH or IgL. BCR “cross-linking” with this reagent mimics the binding of polyvalent antigen and initiates a rapid cascade of well-known proximal phosphorylation events, involving multiple kinases and adaptor molecules including Src family kinases (SFK, chiefly LYN), spleen tyrosine kinase (SYK), Bruton tyrosine kinase (BTK), and PI3K. Active BCR signaling is therefore “druggable” by small-molecule inhibitors (SMIs) of several kinases, potentially preventing the activation of one or more of the distal signaling pathways that drive proliferation, growth, and survival: NF-κB, NFAT, MAPK, and AKT/mTOR.
If active BCR signaling is relevant to B-cell malignancies, a fundamental implication is that the BCR in malignant B cells is specific for an antigen that it encounters in vivo. Most evidence supporting this implication comes from chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), which can be divided into 2 types of cases. In unmutated (U) cases, the variable (V), diversity (D), and joining (J) segments of the IgH and IgL genes that have been selected by recombination to encode HVRs have germline sequences. In mutated (M) cases, these segments have undergone somatic hypermutation, the normal process in germinal centers by which the affinity of BCR for its cognate antigen is increased. Both U and M cases display “stereotyped” nonrandom utilization of V, D, and J segments, and HVRs from some M cases have identical nucleotide sequences, both of which imply selection for binding to a common self-antigen. Although the BCR of CLL cells has often been considered to resemble “natural” polyreactive antibodies characteristic of normal marginal zone B cells, specific antigens recognized by CLL/SLL cells have been identified, mostly of self-origin2,3 but also of fungal origin in some cases.4 A recent study used a cell-based model for ectopic expression of paired IgH and IgL genes derived from various B-cell malignancies; remarkably, spontaneous signaling (as indicated by Ca2+ flux) was observed for BCR derived from all 17 CLL/SLL cases (of both U and M types), but not other B-cell malignancies.5 Further investigation indicated that the CLL/SLL BCR was being activated by binding to an epitope in the IgH chain of the BCR itself, a finding that has been reported by another group involving a different epitope of the IgH chain.6 Approximately 20% of cases of follicular lymphoma (FL) and mantle cell lymphoma (MCL) were found to have nonstereotyped BCRs that bind to a common autoantigen in the N-terminal region of vimentin.7 Another study found evidence for BCR self-reactivity in 25% of cases of FL and identified a specific antigen in one case.8 Activation of the FL BCR has also been proposed to occur by a mechanism other than antigen binding by the HVR: unusually frequent mutations in IgH scaffold regions lead to abnormal sites of N-glycosylation in FL IgH chains,9 which may be bound by microenvironmental lectins and trigger BCR signaling.10 Although an infectious agent is believed to be causal (at least initially) of mucosal-associated lymphoid tissue (MALT) lymphomas occurring in various sites, most notably Helicobacter pylori in gastric MALT lymphomas, BCRs from these lymphomas are not directed against the organism but against one or more self-antigens.11
Active BCR signaling as a model for BCR signaling in B-cell malignancies has additional implications. One is that self-reactivity by B-cell malignancies implies a defect in tolerance mechanisms that normally prevent development of self-reactive B cells. Although tolerance mechanisms are incompletely understood, their disruption is believed to be central to B-cell–mediated autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis and, indeed, the frequency of patients with both autoimmune diseases and B-cell malignancies is significantly higher than expected from coincidence or chance.12,13 The most therapeutically significant implication is that downstream signaling pathways activated by acute BCR signaling should also be active in malignant B cells. This was found to be the case for the activated B-cell (ABC) subtype of diffuse large B-cell lymphoma (DLBCL), originally defined by having a gene expression profile with similarities to that of normal memory B cells activated by acute BCR cross-linking,14 and subsequently found to depend on NF-κB activation by the CARD11/ BCL10/MALT1 complex.15,16 RNA interference studies found that the viability of most ABC-DLBCL cell lines was compromised by knock-down of several signaling proteins downstream of the BCR (BTK, SYK, MALT1, and CARD11), as well as IgH and IgL.17 The most proximal signal transducing elements of the BCR, CD79A and CD79B, which are noncovalently associated with 2 IgH and 2 IgL disulfide bond-linked chains to comprise the BCR, were also found to be required for viability of BCR-dependent ABC-DLBCL cell lines. Sequencing found that most of these lines, and 24% of primary ABC-DLBCL tumors, had mutations in the immunoreceptor tyrosine activation motif (ITAM) domains of CD79A and CD79B genes, which normally serve as substrates for phosphorylation by SFK and activation of SYK via its tandem SH2 domains. The molecular consequences of these ITAM mutations is not entirely clear, although they appear to enhance BCR signaling by promoting surface BCR expression and reducing signal-terminating LYN kinase activity. Nonetheless, the frequency of these BCR-activating mutations in primary tumors is strong evidence for “chronic active” BCR signaling in ABC-DLBCL. Of therapeutic importance, in a separate 10% of ABC-DLBCL tumors, the essential consequences of chronic active BCR signaling appear to be replicated by mutations in the coiled-coil linker domain of CARD11, inducing CARD11 to spontaneously activate NF-κB without BCR input.17,18 Despite the expression of a BCR by cell lines and primary tumors of the germinal center B-cell (GCB) subtype of DLBCL, to date, there is no clear evidence for chronic active BCR signaling in this subtype.
Normal B cells also exhibit “tonic,” antigen-independent BCR signaling that is relevant to B-cell malignancies. After productive IgH rearrangement, early B-cell precursors in the BM express a pre-BCR (consisting of IgH paired with a surrogate light chain, CD79A, and CD79B) that generates antigen-independent, ITAM-transmitted signals promoting subsequent development and survival. In the periphery, mature B cells continue to depend on tonic BCR signaling, as shown by their disappearance upon conditional deletion of IgH or CD79A.19 In contrast, conditional deletion of members of the CARD11/ BCL10/MALT1 complex, essential for acute or chronic active BCR signaling, does not eliminate B cells. Rescue of B cells after BCR deletion was provided by a constitutively active PI3K gene,20 indicating activation of PI3K by tonic BCR signaling and suggesting a therapeutic target for its inhibition. Although both tonic and chronic active BCR signaling may occur in multiple types of B-cell malignancies, Burkitt lymphoma (BL) appears to be the best example of a B-cell malignancy driven by tonic BCR signaling alone. Frequent (70%) mutations in TCF3 or its negative regulator ID3 in BL cell lines and primary tumors were found to be associated with increased expression of IgH and IgL, and RNA interference studies showed dependence on CD79A and SYK but not CARD11.21 Other investigations, and the use of SMIs, indicated dependence on activation of the PI3K/AKT/mTOR pathway in BL (Figure 1).

Targets of BCR signaling. Signaling through the B-cell receptor (BCR) leads to activation of multiple downstream kinases and is critical for cell survival. Agents are in early and late clinical development targeting key components of the cascade, including Bruton tyrosine kinase (BTK), PI3K, SYK, mammalian target of rapamycin (MTOR), and AKT appear promising with activity observed across B-cell malignancies. CK indicates cytokine; TLR, toll-like receptor; Ca, calcium; PIP3, phosphatidylinositol triphosphase; and PIP2, phosphatidylinositol biphosphate.
Targets of BCR signaling. Signaling through the B-cell receptor (BCR) leads to activation of multiple downstream kinases and is critical for cell survival. Agents are in early and late clinical development targeting key components of the cascade, including Bruton tyrosine kinase (BTK), PI3K, SYK, mammalian target of rapamycin (MTOR), and AKT appear promising with activity observed across B-cell malignancies. CK indicates cytokine; TLR, toll-like receptor; Ca, calcium; PIP3, phosphatidylinositol triphosphase; and PIP2, phosphatidylinositol biphosphate.
BTK inhibition
BTK is a non-receptor kinase, the function of which is essential to normal B cells, as shown by the absence of B cells in patients with Bruton agammaglobulinemia, who have inactivating BTK mutations. In acute and chronic active BCR signaling, BTK is phosphorylated by SYK and then phosphorylates phospholipase Cγ2, leading to activation of protein kinase C beta and, in turn, CARD11. Ibrutinib (PCI-32765) is an orally available, selective kinase inhibitor that irreversibly binds to the Cys-481 residue of BTK. Early preclinical studies demonstrated ibrutinib's ability to block BCR signaling in normal peripheral B cells and induce response in animals with lymphoma.22 In the initial phase 1 study of ibrutinib in patients with B-cell malignancies, the side effect profile was mild with minimal myelosuppression, and a maximum tolerated dose was not reached. No cumulative hematologic or nonhematologic toxicity was reported in patients with prolonged dosing. There was no significant reduction in normal B-cell or circulating immunoglobulin levels observed. A fluorescent derivative of PCI-32765 was used to quantify BTK active site occupancy by study drug and showed near-complete target binding at low dose levels. Objective responses were reported in multiple histologies, with significant activity observed in the majority of patients with MCL and CLL; overall response rates (ORR) were 78% and 69%, respectively.23
These preliminary findings prompted multiple phase 2 studies of ibrutinib in relapsed/refractory aggressive and indolent NHL. In a phase 2 single-arm multicenter study in patients with relapsed MCL, early results in 111 patients showed an ORR of 74%, including 35% for complete remission.24 Interestingly, patients had improved response with longer exposure to drug and 53% of patients remain on study with a median time on treatment of 11 months. Improved response with prolonged exposure was also observed in patients with indolent lymphoma, with nearly 12 months of treatment required for complete response in some patients.25 The ORRs were 71% in elderly treatment-naive patients with CLL, including 67% in patients harboring a 17p deletion.26 Progression-free survival (PFS) was 76% and 96% at 22 months in high-risk and relapsed patients, respectively. In relapsed DLBCL, higher responses were observed in 40% of patients with the ABC subtype, as expected, compared with only 5% for GCB subtype patients.27 The mild side effect profile reported from these larger efficacy studies appears to mirror that reported from the initial trials, and several patients remain on ibrutinib beyond 1 year with continued response. The majority of patients with Waldenstrom macroglobulinemia harbor a mutation in MYD88 that may result in aberrant signaling through the BCR pathway. Three of 4 patients in the initial phase 1 study of ibrutinib with Waldenstrom macroglobulinemia attained a partial response and phase 2 studies are now under way.
CC292 and ONO-WG-307 are 2 other BTK inhibitors in clinical development. Preclinical models have demonstrated CC292's ability to form a highly selective covalent bond with the cysteine residue in BTK. Preliminary results from an ongoing phase 1 study in relapsed hematologic malignancies suggest significant activity in CLL and some NHLs.28 Clinical results from trials with ONO-WG-307 have yet to be reported, but the agent appears to reversibly bind BTK, and early studies have demonstrated activity in animal xenograft models of lymphoma.
PI3K inhibition
The oncogenic PI3K/AKT pathway has been well characterized and is critical for essential cellular processes such as metabolism, growth, and proliferation. Activation leads to inhibition of apoptotic signals while concurrently increasing survival signals through activation of NF-κB.29 Although PI3K is broadly expressed in multiple cell types, the p110 delta and p110 gamma isoforms are expressed primarily in cells of hematopoietic origin and therefore represent a promising therapeutic target for lymphoma therapy.30 Inhibition of these isoforms has also been shown to affect the microenvironment through suppression of tumor-associated inflammation, cell signaling, and angiogenesis.31,32
GS1101 is an oral agent that primarily inhibits the delta isoform of PI3K. In phase 1 studies, GS1101 was well tolerated and demonstrated substantial antitumor activity in multiple B-cell malignancies. In the initial dose-finding study, a maximum tolerated dose was not reached within the preplanned dose range, and 61% and 33% of patients with MCL33 and CLL34 demonstrated response. Although only a minority of patients attained an objective response, nearly all patients with CLL experienced a reduction in the size of peripheral adenopathy. As has been observed with ibrutinib therapy, the rapid reduction in lymph node disease was accompanied by circulating lymphocytosis, highlighting the inhibitor's ability to induce a “compartmental shift” of malignant lymphocytes out of the BM and lymph nodes and into the peripheral blood. Recent preclinical studies suggest that this effect may be in part due to changes in the expression and response of cell adhesion/migration proteins. In vitro studies of CLL cells have provided various evidence that SMIs of BTK, PI3K, or SYK can reduce migration in response to chemokines CXCL12 and CXCL13 (produced by CLL-supporting microenvironmental stromal cells), production of CCL3 and CCL4 (T-cell-recruiting chemokines) by CLL cells, and other measures of BCR signaling in CLL cells.32,35-39
IPI-145 is an orally bioavailable inhibitor of PI3K with activity against both gamma and delta isoforms. Preliminary results from early phase 1 studies in hematologic malignancies appear promising, with responses observed in several B-cell subtypes as well as Hodgkins lymphoma and T-cell NHLs. Eighty-two percent of evaluable CLL patients demonstrated a partial or nodal response.40
SYK inhibition
SYK is a proximal and key element of the BCR pathway that is expressed primarily in cells of hematopoietic origin. As with other proximal kinases in the BCR, phosphorylation of SYK leads to the activation and amplification of downstream receptor signaling and likely plays a role in chemokine signaling and cellular responses.41 Fostamatinib is a competitive inhibitor of SYK that has been shown in vitro to inhibit BCR signaling in CLL models. Similar to other inhibitors of the BCR, fostamatinib may also reduce the protective effect of stromal cells in the immune microenvironment.37 In a phase 1/2 study of fostamatinib in patients with relapsed hematologic malignancies, responses were observed in CLL (ORR 54%) as well as MCL, DLBCL, and FL.42 The most common toxicities reported were diarrhea, fatigue, cytopenias, hypertension, and nausea. Phase 2 studies are currently under way in aggressive B-cell NHL. Other SYK inhibitors such as GS-9973 are in development, and studies were recently started investigating the potential role of these agents in multiple histologies (Table 1).
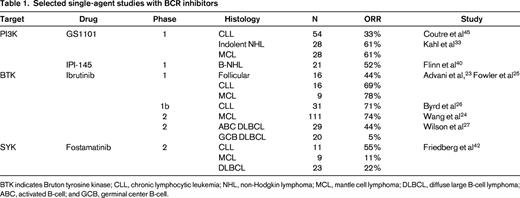
Integration of BCR inhibitors into multiagent therapy regimens
Although efficacy has been observed with multiple inhibitors of the BCR pathway (BCRi's), responses are slow and most patients fail to attain a complete remission. In DLBCL, response is uncommon or transient in most patients, and in indolent disease, many patients still have evidence of peripheral blood and/or BM involvement after extended treatment. The effect that chronic exposure may have on resistance, immunosuppression, and unforeseen long-term toxicity is also unknown. These problems may be addressed by combining BCRi's with other agents, for which the mild side effect profile, oral availability, and non-cross-resistant mechanism of several BCRi agents make them attractive.
One approach is to combine BCRi's with currently available and/or emerging noncytotoxic agents that target alternate pathways. Among preliminary studies combining BCRi's with anti-CD20–directed therapy, Burger et al recently treated 40 patients with high-risk relapsed CLL with ibrutinib plus weekly rituximab for 4 weeks, then monthly rituximab for 5 months.43 In this group with poor prognostic factors (nearly 50% had 17p deletion), the ORR was 85% with no appreciable increase in toxicity with the addition of rituximab. The investigators also observed an earlier rise and peak in WBC count and a shorter duration of lymphocytosis than in single-agent ibrutinib studies. In patients with relapsed indolent NHL, the combination of GS1101 plus rituximab also appears safe and active; GS1101 given twice daily with weekly rituximab for 8 doses demonstrated an ORR of 77% and a 1-year PFS of 82%.44 Using similar dosing in CLL, the ORR was 78% with a 74% PFS at 1 year.45 The efficacy of this approach is being further validated in recently launched phase 3 studies comparing rituximab plus GS1101 versus rituximab alone in patients with relapsed CLL. In preclinical models of CLL, acute BCR signaling induced by anti-IgM reduced the effect of pro-apoptotic agents such as the mitochondrial-targeting agent ABT-737; this effect of anti-IgM was mitigated in the presence of inhibitors of BTK or PI3K, and combining a proapoptotic agent with ibrutinib or the PI3K inhibitor idelalisib was shown to cause a synergistic decline in viable primary CLL cells.46
In ABC-DLBCL, there is strong but still preclinical rationale for combining BCRi's with inhibition of another oncogenic pathway. Gain-of-function mutations in MYD88, found in 35% of primary tumors, cause the associated kinases IRAK1 and IRAK4 to activate NF-κB in a manner distinct from the BCR, similarly promoting IRF4 but uniquely promoting IFN-β production.47 Mutations in MYD88 and BCR pathway-relevant genes have partial overlap with BCR pathway mutations in some tumors and cell lines, and in these lines toxicity is increased by specific inhibition of both pathways. Although intentional inhibitors of the MYD88 pathway are still in development, the impressive single-agent efficacy of lenalidomide against ABC-DLBCL was discovered to depend on its differential modulation of MYD88 effects, further increasing IFNB to toxic levels while blocking the up-regulation of IRF4, and killing of ABC-DLBCL cell lines by lenalidomide was increased by BCRi's.48
Dual targeting of the multiple kinases within the BCR pathway may also increase antitumor activity. In CLL cells cultured with stroma-conditioned media, the combination of a SYK inhibitor (GS-9973) and a PI3K inhibitor (idelalisib) demonstrated synergistic activity and decreased downstream signaling within the BCR pathway.36 Phase 2 combination studies with the 2 agents are under way.
Among studies combining BCRi's with traditional chemotherapy in indolent NHL, rapid reduction in peripheral lymphadenopathy was observed in most patients receiving GS1101 with 90mg/m2 of bendamustine with or without rituximab (ORR% 77%-85%).44 Responses with the regimen were durable at 1 year in most patients (78%-90%) and the addition of GS1101 did not appear to cause overlapping toxicities. In patients with relapsed CLL using a similar combination, a response was observed in 81% of patients with an estimated 2-year PFS of 62%.49 Full-dose bendamustine and rituximab are also being explored with escalating doses of ibrutinib in an ongoing phase 1 study. Although cytopenias were observed, a dose-limiting toxicity was not reached with the combination, and the median number of cycles received was 3 (of 6).50 Preliminary results of studies with ibrutinib plus R-CHOP (rituximab plus cyclophosphamide, hydroxydaunorubicin, vincristine, prednisone/prednisolone) in aggressive B-cell lymphomas were recently reported with high overall and complete responses (100% and 67%, respectively) observed (Table 2).51-53
Selected combination studies with BCR inhibitors
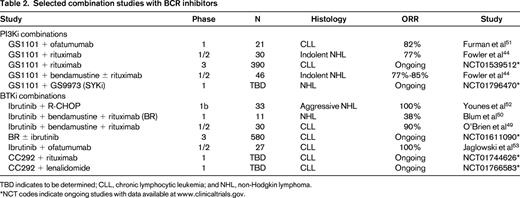
TBD indicates to be determined; CLL, chronic lymphocytic leukemia; and NHL, non-Hodgkin lymphoma.
*NCT codes indicate ongoing studies with data available at www.clinicaltrials.gov.
Further issues in clinical translation of BCR-targeting therapy
Preclinical evidence suggests that BCR signaling is frequently an essential feature in many types of B-cell malignancies, and this appears to have been verified by the promising efficacy of BCRi's in early clinical trials. However, not all patients are initially responsive, even allowing for the slow onset of response that is characteristic of some BCRi's. Despite their favorable side-effect profile, BCRi's should not be given to patients who will not respond if those patients can be identified in advance. There is therefore a need for the development of clinically applicable biomarkers predictive of response to BCRi's. Current efforts in that regard are largely limited to CLIA-compatible assays that can distinguish the ABC and GCB subtypes of DLBCL, but that is likely to be an imperfect predictor of response; the ORR in the phase 2 single-agent trial of ibrutinib in relapsed ABC-DLBCL was only 40%, and there are known molecular abnormalities in untreated ABC-DLBCL tumors (such as CARD11 mutations) that would likely make them BCRi resistant. Within ABC-DLBCL patients in the Phase 2 trial of ibrutinib, the pattern of BCR-relevant mutations (in CD79A, CD79B, and CARD11) showed partial correlation with response,27 but not all BCR-dependent, BCRi-sensitive ABC-DLBCL lines have ITAM mutations without CARD11 mutations. Patients with both CD79B and MYD88 L265P mutations (n = 4) responded, whereas patients with only a MYD88 L265P mutation were resistant. The phosphorylation status of BCR signaling molecules at relevant sites might be a more direct predictor of sensitivity to BCRi's. Blood levels of BCR-driven chemokines (CCL3 and CCL4) are predictive of disease progression in CLL54 and biomarkers of initial response to BCRi's in CLL32,38 ; their role as biomarkers of BCR activation in other B-cell malignancies is currently unknown, but older studies showed that CCL3 mRNA levels in DLBCL tumor biopsies were predictive of shorter survival.55
The development of resistance to BCRi's must be anticipated and has anecdotally been observed in some patients in single-agent trials. This may be lessened by the use of BCRi's as part of combination regimens and/or in the frontline setting but is still of concern. Potential mechanisms of resistance within the BCR signaling pathway include the acquisition of mutations under selection pressure, such as in CARD11 or the ibrutinib-targeting residue of BTK. Less obvious mechanisms of resistance would involve the activation of other pathways that might substitute for the loss of BCR signaling or the inactivation of pathways responsible for the spontaneous cell death caused by BCRi's. Given that up-regulation of IFN-β, driven by gain-of-function mutations in MYD88, is responsible for ABC-DLBCL cell line killing by lenalidomide,48 it is plausible that mutations affecting IFN-β production or response could promote resistance to BCRi's.
Conclusion
The development of specific inhibitors of the BCR pathway represents a major breakthrough in the treatment of B-cell malignancies. These advances were a result of close collaboration between clinical and basic scientists at multiple institutions and highlight the importance and need for pioneering preclinical and mechanistic studies combined with rational and science-based drug design. However, despite early success and substantial and durable responses in some patients, there is still much work to be done. Many patients fail to respond or progress rapidly on treatment and the mechanism(s) of primary or acquired resistance are still unknown. As has been observed with other targeted biologic agents for lymphoma and leukemia, the greatest impact from inhibition of the BCR pathway may come from combination therapy, possibly in the frontline setting. Integrating correlative studies into ongoing and future studies will be essential to answer these key questions and ultimately lead to the development of highly effective mechanism-based drug regimens.
Disclosures
Conflict-of-interest disclosure: N.F. is on the board of directors or an advisory committee for Celgene, Roche, Jannsen, Phamacyclics, and Gillead and has received research funding from Celgene, Roche, Pharmacyclics, and Gillead. E.D. declares no competing financial interests. Off-label drug use: GS1101, ibrutinib, fostamatinib, and IPI-145 are not FDA approved for lymphoma. Early clinical trial results will be discussed.
Correspondence
Nathan Fowler, Department of Lymphoma/Myeloma, MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 429, Houston, TX 77030; Phone: 713-792-2860; Fax: 713-794-5656; e-mail: nfowler@mdanderson.org.
