
Abstract
Clinical experience with 5-azacytidine and decitabine treatment of myelodysplastic syndromes (MDS), complemented by biological and pharmacological studies, has revealed compelling mechanism of action differences compared with traditional myeloid cancer treatment mainstays such as cytarabine. For example, 5-azacytidine and decitabine produce remissions and better overall survival in MDS with high-risk chromosome abnormalities at a surprisingly high rate, consistent with experimental observations that noncytotoxic DNA methyltransferase depletion by 5-azacytidine/decitabine can trigger cell cycle exit independently of p53, thus circumventing a basis for resistance to apoptosis-based DNA-damaging therapy. That responses cut across the chaotic genomic landscape of MDS highlights common threads in disease, such as high expression in myeloblasts of differentiation-driving transcription factors yet paradoxical epigenetic suppression of proliferation-terminating late-differentiation genes. Less toxic regimens (lower dosages but more frequent administration) of 5-azacytidine/decitabine have been more successful, underscoring the importance of preserving functionally normal stem cells, which are rendered more precious by attrition from age, previous cytotoxic treatments, and the disease process and are needed to relieve cytopenias, the cause of morbidity and mortality. Also emphasized is that there can be no therapeutic benefit, regardless of mutation or cytogenetic subtype, if DNA methyltransferase is not depleted by sufficient overlap between intracellular drug half-lives and S-phase entries of malignant cells. Improved understanding of mechanism-of-action differences demands new approaches, from historic (but not scientific) more-is-better and one-size-fits-all empiricism to pharmacodynamic-based designs and combinations directed not solely at suppressing malignant clones, but at improving therapeutic indices.
Introduction
The preceding decade has seen the approval of 2 frontline therapies, 5-azacytidine and decitabine, for all subtypes of myelodysplastic syndromes (MDS) and one frontline therapy, lenalidomide, for MDS containing a chromosome 5q deletion. An ideal treatment would suppress malignant clones but preserve or even promote functionally normal hematopoietic stem and progenitor cells (favorable therapeutic index), because these are crucial to relieve life-threatening cytopenia and may already be depleted by age, previous therapies, and the disease process.1,2 Antimetabolite, DNA-damaging, or cytotoxic treatments for myeloid or other cancers do not approach this ideal because malignant clones often genetically inactivate key apoptosis regulators (eg, TP53). Therefore, apoptosis-based therapy, although often initially efficacious, selects against normal cells in which apoptosis genes are intact while selecting for apoptosis-resistant malignant subclones.3,4 Second- or third-line cytotoxic drugs may have different proximal molecular targets, but because the final common pathway is apoptosis, relapsed disease is refractory. Although cytotoxic treatments have transformed the outlook for certain subsets of myeloid cancer (eg, acute myeloid leukemia [AML] with recurrent genetic abnormalities and intact p53-systems), decades of using aggressive cytotoxic therapy suggest that further escalation in this direction, with even more cytotoxic drugs or combinations, is unlikely to substantially improve outcomes in other myeloid cancers without stem cell transplantation.
Fortunately, malignant growth can be terminated by means other than cytotoxicity. Here, 5 key clinical observations from clinical trials in MDS of the DNA methyl-transferase 1 (DNMT1)–depleting drugs 5-azacytidine and decitabine are described. These observations are platforms from which interactions between these drugs and MDS biology can be viewed with clarity. The mechanistic basis for the strengths and limitations of these drugs exposed to practitioners in this way permits more comfortable and expert use to suppress even genomically complex malignant clones while sparing the normal stem cells needed to relieve cytopenia. Real-world situations and potential mechanism-based solutions are described in the “How I treat” section, followed by a discussion of logical next steps that can be taken to advance meaningfully in this new direction.
Clinical observation #1
5-Azacytidine/decitabine therapy produces complete cytogenetic remissions (Cyto-CRs) even in MDS containing high-risk chromosome abnormalities
In patients with chromosome 7 deletions known to imply high risks, overall survival (OS) was 13.1 months in patients treated with 5-azacytidine versus 4.6 months in patients randomized to conventional care.5 In several clinical trials with decitabine, Cyto-CR rates have ranged between 35% and 50%, even in patients with complex (≥ 3) chromosome abnormalities5-10 (historic response rates with cytarabine-based therapy are described below). Cyto-CR rates of 50%, including patients with complex abnormalities and TP53 mutation, have also been seen when decitabine was administered using a regimen explicitly designed to avoid cytotoxic effects.11
Mechanisms and lessons #1
Mutations and deletions in the p53-system confer resistance to multiple therapeutics.
Conventional chemotherapeutics to treat AML have differing proximal mechanisms of action, such as topoisomerase inhibition (daunorubicin) or termination of DNA chain synthesis (cytarabine); however, a final common pathway converges onto p53, a stress and DNA damage sensor that together with p16/CDKN2A is a master regulator of cell cycle exit during apoptosis (ie, cytotoxicity; for review, see Saunthararajah et al3 ). Therefore, not surprisingly, mutation and chromosome deletion at the TP53 locus is associated with resistance to conventional chemotherapy: partial remission or CR with intensive chemotherapy was achieved in 81% of AML cases without and in 33% of cases with TP53 mutations.12 Similarly, responses in MDS patients treated with intensive chemotherapy or low-dose cytarabine were 60% of cases without and 8% of cases with TP53 mutations.12 The rate of TP53 mutation can exceeded 70% in MDS and AML cases with complex cytogenetic abnormalities. Even when TP53 itself is not directly mutated or deleted, the p53 system is frequently attenuated by genetic abnormalities in p53 cofactors: TP53 defects or MDM4 gain were noted in 45.5% of patients with blast transformed myeloproliferative disease, which responds only transiently if at all to conventional cytotoxic therapy.13
Cell cycle exit by differentiation does not require p53.
These limitations of cytotoxic therapy give rise to the question, does a p53-independent mechanism of action explain the effectiveness of 5-azacytidine/decitabine in MDS with high-risk chromosome abnormalities? Although p53-null mice are cancer prone, the development of these mice is essentially normal, with normal patterns of differentiation in almost all tissues. In other words, differentiation-mediated cell cycle exit does not require p53. Numerous studies have described terminal differentiation of AML cells, including p53-null cells, treated with conditions or drugs that inhibit chromatin-modifying enzymes, such as 5-azacytidine and decitabine that deplete DNMT1 (for review, see Saunthararajah et al3 ). However, high concentrations of 5-azacytidine and decitabine have antimetabolite and DNA-damaging effects and can trigger apoptosis. Therefore, for many years, there has been controversy as to whether differentiation-mediated cell cycle exit is the most important clinical action of 5-azacytidine or decitabine (for review, see Tuma et al14 ). There has even been debate about whether chromatin-modifying enzyme inhibition is the most important effect of these drugs.14 Recently, however, preclinical in vitro and in vivo studies followed by a proof-of-concept clinical trial, have demonstrated that DNA damage and apoptosis are not necessary for decitabine-induced cell cycle exit of myeloid cancer cells11,15-19 (DNMT1 depletion by 5-azacytidine/decitabine can be separated from cytotoxicity because, unlike most other nucleoside analogs, the sugar moieties of these drugs are physiologic and do not terminate chain elongation after incorporation into DNA3 ). Resolving the controversy of whether noncytotoxic doses can be therapeutic was important because it justifies optimizing dosage to achieve the specific molecular pharmacodynamic effect of interest, DNMT1 depletion. Quite possibly, cytotoxic effects can be counterproductive by destroying functionally normal stem cells and causing toxicity that limits the frequency of drug administration, which for mechanism-of-action reasons discussed next, is a critical determinant of efficacy.15-18
“Low-dose” decitabine regimens: not low-dose enough?
Consistent with the idea that an optimum biologic dose concept better serves 5-azacytidine and decitabine than the “maximum-tolerated dose” basis usually used to guide application of nucleoside analogs, a decrease in the decitabine daily dose to 20 from 45 mg/m2 and an increase in the frequency of administration to 5 days every 28 days from 3 days every 42 days, produced a 2- to 3-fold improvement in both MDS remission and hematologic improvement rates.7,9 The lesson here was that lowering the dosage, which should decrease cytotoxic effects, and administration of this lower dosage more often, which should increase S-phase-dependent DNMT1 depletion, is better. Perhaps the dosage should only be as much as is needed to deplete DNMT1, and then the decrease in toxicity can be used to administer the drug even more often to increase the number of malignant cells exposed to S-phase-dependent DNMT1 depletion. A useful analogy is to the game of “Whac-a-Mole”: using a big mallet infrequently hits few moles unnecessarily hard; hitting more frequently, in a distributed fashion, and with a mallet of sufficient but not excessive size, would be more efficient and less damaging to the landscape. By this logic, there are indicators from clinical experience with both decitabine and 5-azacytidine that the current Food and Drug Administration (FDA)–approved dosages of decitabine could be counterproductively high.
Clinical trials that have evaluated decitabine as a treatment for the hemoglobinopathies (nonmalignant diseases in which the need to avoid DNA damage is clear) have demonstrated that dosages of 3.5-7 mg/m2 are sufficient to produce a therapeutic epigenetic effect in vivo, can be administered up to 3 times/wk, and have potent effects on hematopoietic differentiation without cytotoxicity.20,21 Therefore, to continue the logical progression toward minimal nontoxic dosages required to deplete DNMT1, and exploiting the decrease in toxicity to administer drug more often (“Whac-a-Mole”), a clinical trial was conducted in MDS using decitabine 0.1-0.2 mg/kg (∼ 3.5-7 mg/m2) administered 1-3 times/wk subcutaneously. Complete hematologic and cytogenetic remissions were produced, even in high-risk disease with complex chromosome abnormalities, with an overall response rate of 44%.11 As per the studies of the same decitabine regimen in hemoglobinopathies, there was no scientific correlative evidence of cytotoxicity.11,20,21
In a story line similar to that of decitabine, FDA-approved and typically administered 5-azacytidine dosages of 50-75 mg/m2 for 5-10 days every 28 days are 2- to 10-fold lower than the maximum-tolerated dose–derived 5-azacytidine dosages used in the earliest clinical trials.22 How are 5-azacytidine and decitabine related? In vivo, after phosphorylation by uridine cytidine kinases, 5-azacytidine is converted into a corresponding diphosphate that is then converted into decitabine diphosphate by ribonucleotide reductase. The net effect is that ∼ 10% of the ribonucleoside 5-azacytidine is converted into a deoxyribonucleoside triphosphate (ie, decitabine triphosphate) that can be incorporated into DNA, wherein it can deplete DNMT1. Therefore, with respect to DNMT1 depletion, 5-azacytidine is a prodrug of decitabine triphosphate and the FDA-approved dose of 5-azacytidine is approximately equivalent to a decitabine dosage of 7.5 mg/m2. With the important caveat that there have been no head-to-head comparisons, the overall impression has been that the FDA-approved regimen of 5-azacytidine is associated with less neutropenic fever and better efficacy than the FDA-approved regimens of decitabine (20-45 mg/m2/d for 3-5 days every 4-6 weeks; for review, see Gore23 ; Tables 1, 2). Therefore, experience with both 5-azacytidine and decitabine suggest that dosages of decitabine much lower than the FDA-approved daily dosages are sufficient for the pharmacodynamic effect of DNMT1 depletion, although there are pharmacogenetic differences (discussed later) that could have a role in individualizing this dose.
Summary of results of clinical trials of 5-azacytidine in MDS
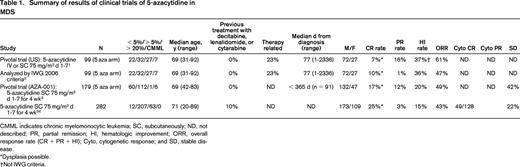
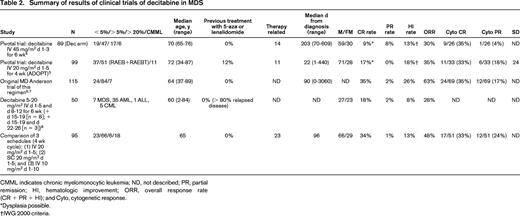
CMML indicates chronic myelomonocytic leukemia; SC, subcutaneously; ND, not described; PR, partial remission; HI, hematologic improvement; ORR, overall response rate (CR + PR + HI); Cyto, cytogenetic response; and SD, stable disease.
*Dysplasia possible.
†Not IWG criteria.
Why do these treatments induce cell cycle exit in MDS/AML cells by p53-independent mechanisms?
Experimental observations, documented by numerous groups over decades, have demonstrated that AML cells exposed to drugs or conditions that inhibit corepressor function (chromatin-“relaxing” conditions) exit the cell cycle by differentiation (for review, see Saunthararajah et al3 ). Recently, the reasons for this have become clear. Chromatin-relaxation per se does not determine gene activation or cell fate: chromatin relaxation in a theoretical cell bereft of DNA-binding transcription factors, which are needed to recruit RNA polymerase and initiate transcription, would not change much if at all; in other words, the baseline expression pattern of transcription factors is a key determinant of the cell fate response to chromatin relaxation.24 Furthermore, not all transcription factors are equal: lineage conversion of mature cells of one lineage into mature cells of distant lineages or into embryonic stem cells by forced expression of as few as 2 specific transcription factors illustrates this.25 What, then, are the master transcription factors that are expressed at high levels in AML cells? AML cells, including demonstrably self-renewing subsets (leukemia initiating/stem cells), express strikingly high levels of key hematopoietic lineage-specifying transcription factors (eg, CEBPA, PU.1/SPI1, and GATA1).3,15,16 Another notable clinical observation is that the evolution of MDS and myeloproliferative neoplasms into AML is associated with inactivating mutations in key polycomb repressor complex 2 components (eg, EZH2, ASXL1, and DNMT3A) that have an evolutionarily conserved role in suppressing lineage programs.16,26 These and other molecular observations indicate that neoplastic evolution of myeloid cancers is not “de-differentiation,” but rather is characterized by further lineage restriction.16,26 There is another translationally important implication of this disease biology: the same chromatin-relaxing treatments that trigger terminal differentiation of the myeloid cancer cells preserve self-renewal of uncommitted normal hematopoietic stem cells, because these cells express stem cell transcription factors, not high levels of lineage-specifying transcription factors.24,27 This seems almost too good to be true, but indeed it is a reproducible observation (for review, see Saunthararajah et al3 ).
Despite higher responses in MDS with high-risk cytogenetics treated with 5-azacytidine/decitabine than with conventional cytotoxic therapy, these patients continue to have relatively poor long-term outcomes. Therefore, the p53-independent concept of treatment appears sound, but nonetheless needs ongoing efforts at optimization.
Clinical observation #2 (2 related observations)
Cytogenetic response rates to 5-azacytidine or decitabine exceed the hematologic response rate
Despite a Cyto-CR rate of 9/26 (35%), a substantially lower complete hematologic remission rate of only 8/89 (9%) was observed with decitabine administered for 3 days every 6 weeks.6 Similarly, with decitabine administered for 5 days every 4 weeks,7 the Cyto-CR rate was 11/33 (33%), whereas the complete hematologic remission rate was only 17/99 (17%). A similar disconnect between cytogenetic and hematologic response rates is observed with 5-azacytidine.28
Progressive disease often occurs after a period of time (median 4 months) in which 5-azacytidine or decitabine have not been administered because of cytopenia
There is a frequent sequence of events in patients who lose response on 5-azacytidine or decitabine therapy: first, there is progressive cytopenia; next, BM examination demonstrates hypocellularity without an increase in myeloblasts as the cause of cytopenia, so treatment is held; and finally, after months without therapy (median 4 months29 ), there is frank progressive disease.29
Mechanisms and lessons #2
Cytogenetic response and stable disease are produced by suppression of malignant clones.
As discussed earlier, cell cycle exit by p53-independent mechanisms explains the substantial efficacy of these drugs in suppressing karyotypically abnormal malignant clones that are resistant to conventional apoptosis-based therapy. Also supporting major efficacy in suppressing malignant clones, stable disease, another indicator of at least partial malignant clone suppression (stable disease per International Working Group [IWG] criteria is associated with better survival than progressive disease),30 or better was observed in 91% of patients treated in the AZA-001 trial of 5-azacytidine.5
Hematologic response depends not only on suppression of malignant clones, but also on recovery of functionally normal hematopoietic stem cells.
Such normal cells are diminished by older age, previous DNA-damaging treatments, and the type and duration of the disease process (for review, see Van Zant and Liang1 and Gadalla and Savage2 ). Therefore, attrition of these crucial cells could be a major contributing factor to persistent or recurrent cytopenia. Indeed, there is a subset of MDS patients in which contraction of the normal hematopoietic stem cell pool, rather than outgrowth of a malignant clone with a primary growth advantage, is the primary pathophysiology. These patients are discussed next.
Clonal hematopoiesis as “last man standing.”
Aplastic anemia is a BM failure state in which immune-mediated mechanisms produce BM hypocellularity and peripheral cytopenias. Of patients diagnosed with aplastic anemia and treated with antithymocyte globulin and cyclosporine, approximately 25% eventually develop a clonal myeloid disorder: MDS, AML, and/or paroxysmal nocturnal hemoglobinuria. This clinical overlap, and molecular data including studies linking shortening of telomere length with evolution into MDS/paroxysmal nocturnal hemoglobinuria,2 suggest that clonal hematopoiesis can emerge as a “last-man-standing” phenomenon: an abnormal clone survives an abnormal microenvironment that suppresses the competition (contracts the normal stem cell pool) without necessarily possessing an intrinsic growth advantage. This interpretation is supported by other clinical observations: the HLA-DR15 genetic marker that is strongly linked with aplastic anemia is associated with low-risk MDS only, not with MDS with an increase in myeloblasts.31 Logically, in last-man-standing MDS, suppression of clonal hematopoiesis by drugs such as 5-azacytidine or decitabine may not relieve cytopenia because the microenvironmental abnormality will continue to prevent functional hematopoiesis.32
Clinical observation #3
Hematologic improvement without CR improves OS
A link between CR and longer survival is well accepted in oncology practice. The experience with 5-azacytidine and decitabine, however, has demonstrated that relief of cytopenia, even if technical criteria for CR are unmet, is also linked with longer survival: in an analysis of decitabine-treated patients (N = 31), hematologic improvement sans CR was associated with significantly better OS33 and, in a multicenter cohort study of high-risk MDS patients (N = 282), achievement of any type of hematologic improvement was accompanied by a significant improvement in OS, in a multivariate analysis that incorporated cytogenetic status, myeloblast percentage, and other risk factors.34
Mechanisms and lessons #3
Cytopenia is the cause of morbidity and mortality.
The majority of patients with MDS will die from complications related to cytopenia: infection, hemorrhage, or cardiovascular death that at least in part is related to anemia.35 This underscores again that, conceptually, the focus of therapy should be on combinations or regimens to relieve cytopenia. This requires not a single-minded focus on destruction of malignant clones, but a holistic consideration also toward preservation or promotion of functionally normal stem cells.
Each cycle of therapy treats only a fraction of the malignant clone.
Because the epigenetic therapeutic effects of 5-azacytidine and decitabine are S-phase dependent, each cycle of therapy can only affect the fraction of the malignant clone that enters S-phase in a small window of time during which intracellular levels of decitabine triphosphate meet minimum levels required to substantially deplete DNMT1. Not surprisingly, best responses can occur after as many as 12 cycles of therapy, with a median of 3-3.5 cycles (for review, see Gore23 ). Hematologic improvement without CR, however, illustrates that restoration of more functional hematopoiesis does not require complete eradication of the malignant clone, especially if lack of toxicity facilitates ongoing therapy to continue suppression of the malignant clone. Experience with these drugs demonstrates that this long time-horizon approach can yield better OS than approaches based on aggressive but short-term malignant clone suppression.
Clinical observation #4
Responses to decitabine can occur in MDS that is resistant to 5-azacytidine and vice-versa
CRs with decitabine were reported in 3 of 14 patients with progressive disease, lack of response, or intolerance to 5-azacytidine.36 Responses by IWG criteria were observed in 6/10 patients treated with 5-azacytidine after decitabine and in 7/21 patients treated with decitabine after 5-azacytidine.37
Mechanisms and lessons #4
An important difference in the pharmacology of decitabine and 5-azacytidine.
Although patient numbers were small, these responses highlight that relapse/resistance may not reflect outgrowth of malignant clones that withstand DNMT1 depletion, but outgrowth of malignant cells that have never experienced the pharmacodynamic effect of DNMT1 depletion in the first place. DNMT1 depletion requires S-phase entry overlap with the minimum intracellular level of decitabine triphosphate needed to produce substantial DNMT1 depletion. Therefore, the fraction of the malignant clone in S-phase and the intracellular half-life of the drug are crucial predictors of drug effect, indeed, more so than histology or genetic complexity (Figure 1). Critical determinants of intracellular half-lives are different for decitabine versus 5-azacytidine: decitabine accumulates in cells because phosphorylation by deoxycytidine kinase (DCK) traps the drug in cells (DCK also determines intracellular accumulation of cytarabine). In contrast, 5-azacytidine, a ribonucleoside, accumulates in cells because of phosphorylation by a uridine-cytidine kinase (UCK1 or UCK2), followed by conversion to phosphorylated decitabine by ribonucleotide reductase (Figure 1). The activity of these 2 different enzyme pathways appears to be inversely correlated, depending on cellular emphasis on the salvage or de novo pathways of deoxynucleotide synthesis (Figure 2). Accordingly, resistance by UCK down-regulation and 5-azacytidine intracellular half-life loss does not confer cross-resistance to DCK-dependent decitabine.38
![Figure 1. DCK and UCK enzymes predict drug sensitivity. In cell lines representative of the histologic and genetic diversity of cancer (NCI60), the rate-limiting enzymes that determine the intracellular half-lives of decitabine and 5-azacytidine, DCK and UCK respectively, predicted drug sensitivity. Drug sensitivity is represented as GI50: the drug concentration causing 50% growth reduction. The higher the GI50, the less sensitive to drug, quantified by measurement of total protein at day 6 (raw data downloaded from the National Cancer Institute [http://dtp.nci.nih.gov] and GSE5846, gene expression by microarray). (A) Decitabine GI50 is inversely correlated with DCK expression and growth fraction (higher DCK implies longer intracellular half-life and a higher doubling time implies a smaller S-phase fraction). Decitabine GI50 did not correlate with UCK2 expression (data not shown). (B) 5-azacytidine GI50 inversely correlates with UCK2 expression and growth fraction. 5-Azacytidine GI50 did not correlate with DCK expression (data not shown). UCK1 expression was not available in this database.](/view-large/figure/6501302/bep0011303400001.jpeg)
DCK and UCK enzymes predict drug sensitivity. In cell lines representative of the histologic and genetic diversity of cancer (NCI60), the rate-limiting enzymes that determine the intracellular half-lives of decitabine and 5-azacytidine, DCK and UCK respectively, predicted drug sensitivity. Drug sensitivity is represented as GI50: the drug concentration causing 50% growth reduction. The higher the GI50, the less sensitive to drug, quantified by measurement of total protein at day 6 (raw data downloaded from the National Cancer Institute [http://dtp.nci.nih.gov] and GSE5846, gene expression by microarray). (A) Decitabine GI50 is inversely correlated with DCK expression and growth fraction (higher DCK implies longer intracellular half-life and a higher doubling time implies a smaller S-phase fraction). Decitabine GI50 did not correlate with UCK2 expression (data not shown). (B) 5-azacytidine GI50 inversely correlates with UCK2 expression and growth fraction. 5-Azacytidine GI50 did not correlate with DCK expression (data not shown). UCK1 expression was not available in this database.
DCK and UCK enzymes predict drug sensitivity. In cell lines representative of the histologic and genetic diversity of cancer (NCI60), the rate-limiting enzymes that determine the intracellular half-lives of decitabine and 5-azacytidine, DCK and UCK respectively, predicted drug sensitivity. Drug sensitivity is represented as GI50: the drug concentration causing 50% growth reduction. The higher the GI50, the less sensitive to drug, quantified by measurement of total protein at day 6 (raw data downloaded from the National Cancer Institute [http://dtp.nci.nih.gov] and GSE5846, gene expression by microarray). (A) Decitabine GI50 is inversely correlated with DCK expression and growth fraction (higher DCK implies longer intracellular half-life and a higher doubling time implies a smaller S-phase fraction). Decitabine GI50 did not correlate with UCK2 expression (data not shown). (B) 5-azacytidine GI50 inversely correlates with UCK2 expression and growth fraction. 5-Azacytidine GI50 did not correlate with DCK expression (data not shown). UCK1 expression was not available in this database.

Expression levels of UCK and DCK. Expression levels of UCK and DCK inversely correlate in primary AML cells (n = 178, gene expression measured by RNA sequencing, raw data from The Cancer Genome Atlas). (A) Inverse correlation in DCK and UCK1 expression. (B) Inverse correlation in DCK and UCK2 expression.
Expression levels of UCK and DCK. Expression levels of UCK and DCK inversely correlate in primary AML cells (n = 178, gene expression measured by RNA sequencing, raw data from The Cancer Genome Atlas). (A) Inverse correlation in DCK and UCK1 expression. (B) Inverse correlation in DCK and UCK2 expression.
Clinical observation #5 (2 related observations)
The largest detrimental impact of fewer days of treatment exposure with decitabine was in low-risk (more indolent) MDS
In clinical trials from the same institution, administration of decitabine on a greater number of days (10-20 mg/m2 for 5-10 days every 4 weeks) produced a response rate of 50% in the lowest-risk MDS category and 28% in the higher-risk categories (intermediate-2 and high), whereas administration on fewer days (45 mg/m2/d for 3 days every 6 weeks) produced a response rate of 14% in the lowest-risk MDS category and a response rate of 18% in the higher-risk categories.6,10
Response rates in females may be higher than in males
In a multivariate retrospective analysis that incorporated age, cytogenetics, and myeloblast percentage, the OS of MDS patients treated with 5-azacytidine or decitabine was significantly worse in males (n = 69, median 563 days) compared with females (n = 21, median 1033 days).39 Significantly worse OS in males has also been observed in a cohort of patients with therapy-related MDS (n = 54, median OS 7 months vs 11.1 months in females).40
Mechanisms and lessons #5
Disease kinetics and pharmacodynamic effect.
Even a short treatment exposure may be an effective treatment for a high S-phase fraction malignant disease because the majority of cells may enter the S-phase in the treatment window. Conversely, in disease with a low S-phase fraction, short exposure time may only treat a minor portion of the malignant cells.39
Pharmacogenetic factors and pharmacodynamic effect.
Key enzyme determinants of intracellular half-lives were discussed earlier. What about extracellular half-lives that precede intracellular half-lives? The key determinant of extracellular half-lives of both 5-azacytidine and decitabine is cytidine deaminase (CDA), an enzyme that rapidly deaminates these drugs to uracil base moiety counterparts. The clinical relevance of CDA is illustrated as follows: the half-life of decitabine in buffer in vitro at 37°C is > 10 hours; in contrast, the half-life in vivo is < 10 minutes, a drastic reduction largely attributable to CDA. CDA expression and enzyme activity are significantly higher in males and thus could contribute to the decreased efficacy of these drugs in males, especially in low-risk disease for the reasons discussed above.39,41 Therefore, interindividual variation in disease S-phase fraction and in drug metabolism are 2 reasons to individualize drug dosage and schedule by measurements of pharmacodynamic effect and response.
How I treat
For obvious reasons, hematologists-oncologists are conditioned to suppress malignant clones. In approaching MDS, however, this mindset must be slightly modified. Suppression of malignant clones is a means to an end, not the end itself. The overall purpose of therapy is to halt and reverse a decline in one or more blood counts, and a contraction in the functional hematopoietic stem cell pool could well be contributory.1,2 More thought needs to be given to therapeutic index to spare these valuable commodities. Fortunately, 5-azacytidine and decitabine, which are FDA approved for all subtypes of MDS, facilitate such consideration. Equipped with an appreciation of their mechanism of action, the individual practitioner can comfortably and effectively apply these drugs.
Patient selection and other treatments
Even noncytotoxic therapy that aims to suppress aggressive malignant clones may only exacerbate cytopenia in the subset of MDS patients in whom the primary pathophysiology is microenvironmental alterations (eg, immune-mediated BM failure) or damage that has contracted the normal stem cell pool (last-man-standing MDS1,2 ; Mechanisms #2). Primary immune-mediated pathophysiology should be considered in younger patients with morphologic MDS without an increase in myeloblasts in whom paroxysmal nocturnal hemoglobinuria clones can be detected or who are positive for HLA-DR15,31 and alternative treatments to consider in these patients include aplastic anemia type immunomodulation. There might also be ethnic predispositions to immune-mediated BM failure manifesting as either aplastic anemia or low-risk MDS: aplastic anemia in British Columbia (Canada) is ∼ 4-fold more common among individuals of Asian descent compared with others42 (presumably the environmental influences are similar among the different ethnic groups in Canada). Further, in east Asian countries, there is more MDS without an increase in myeloblasts occurring in younger individuals, whereas in the west, there is more MDS with increased myeloblasts occurring in older individuals.43,44 These biomarkers should not be used as absolute determinants of immunomodulation versus clonal suppression therapy for a particular MDS case: it is most important to recognize that these different primary biologies of disease can operate, and are associated with, particular demographic patterns of disease. Armed with this understanding of pathophysiology, practitioners can better assess the likelihood of aggressive clonal versus last-man-standing MDS and select therapy accordingly.
There are clinical trial data to support the use of thrombopoietin analogs (eg, eltrombopag, romiplostim) to treat immune-mediated BM failure45,46 (the thrombopoietin receptor is present on hematopoietic stem cells). Unfortunately, these drugs (like many new hematology-oncology drugs) are prohibitively expensive and the lack of FDA approval for this indication may prevent their lifesaving application in such patients. There is a concern that these drugs could equally promote malignant clones; perhaps one method to address this concern is to use combinations with 5-azacytidine or decitabine (see “Next steps”).47
For thorough reviews regarding the role of other cytokines (eg, erythropoietin), vitamins, and lenalidomide, please see another recent “How I treat” article.48 Stem cell transplantation remains the only curative treatment option for MDS and should be considered in eligible patients.
In MDS patients who do have aggressive clonal disease, the broad pattern of response suggests that patient selection based on particular mutations or chromosome abnormalities may not be as important as careful attention to 5-azacytidine or decitabine dose and schedule to ensure that it is optimal for individual S-phase fractions and pharmacogenetics (Figures 1, 2, Mechanisms #5, “Next steps”).
Dose
5-Azacytidine.
The historical limits of cytotoxicity, the favorable therapeutic index of the epigenetic effects of 5-azacytidine/decitabine and the evolution of their clinical regimens in this direction create a justification for focusing dosage selection on the need to produce an epigenetic therapeutic effect, rather than antimetabolite cytotoxic effects (Mechanisms #1). 5-Azacytidine dosages of between 50 and 75 mg/m2, administered subcutaneously or infused over 40 minutes, appear to serve this purpose. As discussed in the “Schedule” section below, exposure time is an important determinant of therapeutic effect; therefore, I readily consider dosage reductions to 50 mg/m2 if this will reduce any toxicity and thereby facilitate on-schedule drug administration.
Decitabine.
FDA-approved decitabine dosages of 20-45 mg/m2/d stray into cytotoxic territory. Therefore, in my practice, supported by clinical trial data and experience,11,49,50 I consider dosage reductions of decitabine to as low as 5 mg/m2 to decrease toxicity while preserving a possibility of a therapeutic epigenetic effect; the decrease in toxicity is used to facilitate adherence to schedules of administration.
Schedule
On-time drug administration and persistence.
It is worth reiterating that each cycle of therapy can only affect a fraction of the malignant clone, that which enters S-phase in a small window during which intracellular levels of decitabine-triphosphate meet minimum levels required to substantially deplete DNMT1. Not surprisingly, best responses can occur after as many as 12 cycles of therapy (median 3-3.5 cycles23 ) and discontinuation of therapy is associated with loss of response and disease progression that can occur within 6 months29,51 (Mechanisms #3). In other words, expectations are different from AML induction chemotherapy, in which one expects major suppression of the malignant clone after 1 cycle of therapy. Therefore, every effort should be made to maintain and persist with on-time administration of cycles of therapy so that incremental suppressions of the malignant clone have an opportunity to accumulate.
Use of G-CSF.
In the initial clinical trials of decitabine 20 mg/m2 IV over 1 hour on days 1-5 in 28-day cycles, on-time administration of cycles was emphasized, with G-CSF support if there was a danger that neutropenia would deter on-time administration of drug. In the subsequent multicenter ADOPT study of this regimen, dose reductions were not permitted and delays in cycle administration were used instead; the ADOPT investigators have suggested that this is one potential explanation for a lower response rate than in the earlier study.7 Therefore, I use G-CSF after 5-azacytidine or decitabine if there is a risk that neutropenia and its complications will deter on-time administration of drugs.
Number of days of drug administration per cycle.
A study suggested that 5, 7, or 10 days of 5-azacytidine administration per 28-day cycle were equivalent, but that study was not powered to detect a significant difference between schedules.52 In contrast, first principles (Figure 1) and the overall historical migration from infrequent administration of large dosages to more frequent administration of lower dosages strongly suggest that exposure time (frequency of drug administration) is a critical determinant of the therapeutic effect (Mechanisms #1). In my practice, I do not favor less frequent administration (eg, 5 instead of 7 days of 5-azacytidine per 28-day cycle) and I do consider increasing the number of days of administration of 5-azacytidine 50 mg/m2 to 10 days if malignant clone suppression is inadequate (this is supported by the E1905 US intergroup study28 ). As described in the “Next steps” section, we are actively examining alternative schedules of administration of decitabine.
Consecutive days of administration.
The objective of therapy is not to produce cumulative drug levels that enhance cytotoxicity. Therefore, permitting the weekend to interrupt 7- or 10-day drug administration schedules is not in principle discouraged and has not been shown to be detrimental.
It should be noted that an objective of treatment is to suppress the malignant clone (even if it is by noncytotoxic means11 ). Therefore, nadiring of counts is to be expected, with nadirs approximately 2 weeks into each cycle and lowest in cycle 2 or later.
Loss of response and progressive disease
Hypocellular BM.
As discussed earlier, there is a frequent sequence of events in patients receiving therapy: first, there is progressive cytopenia; BM examination to evaluate cause of cytopenia demonstrates BM hypocellularity without an increase in myeloblasts, so treatment is held. Then, after months without therapy (median 4 months29 ), there is frank progressive disease.29 In other words, the malignant clone is not necessarily growing through 5-azacytidine/decitabine, but instead recovers in the absence of the drug, which is being withheld because of persistent cytopenias because functional hematopoietic stem cells, having undergone attrition by age and other factors, did not recover despite malignant clone suppression1,2 (Mechanisms #2). This is a very difficult, “Next steps” problem that needs thoughtful solutions such as the incorporation into treatment regimens of thrombopoietin analogs that can boost hematopoietic stem cells.45-47
Hypercellular BM.
Another frequent scenario on therapy is frank progressive disease: worsening cytopenia, together with increasing BM cellularity, BM myeloblasts, and/or peripheral myeloblasts. It should be realized that, given the small windows available for interaction between intracellular half-lives and S-phase entries, such progressive disease could be driven by cells that have not been exposed to a treatment effect. Therefore, one consideration is whether schedules of administration are optimal and if an increase in the number of days of administration could be helpful (see “Schedule” section). Another consideration, based on clinical reports and mechanistic reasons, is a switch to the other DNMT1-depleting drug (Mechanisms #4). Clearly, clinical trials with novel agents should be considered (especially if, like 5-azacytidine or decitabine, there are mechanistic reasons to support a p53-independent favorable therapeutic index).
Next steps
Therefore, 5-azacytidine and decitabine are laying out a new path in MDS therapy. Given that most regimen design has been empiric, there are clear opportunities and a need (Figure 3) for rational, mechanism-based improvements in the application of these existing agents in addition to the development of new drugs to advance in this direction.

Survival of MDS patients, defined by ICD-O3 codes 9980 to 9989, at different ages at diagnoses (Surveillance Epidemiology and End Results data). Cases diagnosed since January 1, 2004 are shown in bold (5-azacytidine was FDA approved in 2004); apparent improvements for females in the older age group are marginally significant at P = .06 (log-rank test). Pooling across all years of diagnoses and sex differences in survival are highly significant (P ≪ .0001 left, P = .0001 right, log-rank test; males 2000-2003, n = 1378, ≥ 2004 n = 4021; females 2000-2003 n =1041, ≥ 2004, n = 3144).
Survival of MDS patients, defined by ICD-O3 codes 9980 to 9989, at different ages at diagnoses (Surveillance Epidemiology and End Results data). Cases diagnosed since January 1, 2004 are shown in bold (5-azacytidine was FDA approved in 2004); apparent improvements for females in the older age group are marginally significant at P = .06 (log-rank test). Pooling across all years of diagnoses and sex differences in survival are highly significant (P ≪ .0001 left, P = .0001 right, log-rank test; males 2000-2003, n = 1378, ≥ 2004 n = 4021; females 2000-2003 n =1041, ≥ 2004, n = 3144).
Dose
As for any molecularly targeted treatment, there is a need for practical, peripheral blood pharmacodynamic assays to individualize dosage (to adapt to differences in pharmacogenetics and disease kinetics and to more safely and effectively achieve noncytotoxic DNMT1 depletion). Several groups are pursuing such potential assays.
Schedule
Pulse-cycled schedules of 5-azacytidine/decitabine (intense treatment followed by extended periods of rest) approved by the FDA for MDS treatment have a major limitation: only MDS cells that enter the S-phase during the relatively brief window of treatment exposure are treated. Clonal cells that enter the S-phase during the multiweek intervals between treatment pulses are not exposed to DNMT1 depletion. Therefore, to treat as large a fraction of the MDS clone as possible, more logical schedules might distribute administration of drug (intermittent but frequent) to capture MDS cells entering S-phase asynchronously at different time points. Such an approach would be facilitated by oral formulations of 5-azacytidine and decitabine, both of which are in clinical trials.53,54 Fixed schedules of administration that ignore disease kinetics that vary between individuals or within the same individual over time are clearly overly simplistic: schedules of administration should be modified according to response. Clinical experience with continuous infusions suggests that these might be too toxic, presumably because of excessive intracellular accumulation of drug in normal cells. We and others are examining distributed schedules of administration of decitabine and 5-azacytidine in clinical trials.11,53,54
Route of administration
The pharmacologic profile goal is to extend the window for S-phase-dependent depletion of DNMT1 while also keeping Cmax as low as possible to avoid off-target cytotoxicity (which, by harming normal cells, limits the frequency of administration and, by selecting for the most apoptosis-resistant malignant cells, is counterproductive). Subcutaneous or oral routes of administration are preferable for this purpose; accessibility and long-term treatment considerations favor the oral route.53,54
Combination therapy
Malignant cells must duplicate their deoxycytidines via either salvage or de novo pyrimidine synthesis (Figure 2); decitabine and 5-azacytidine are incorporated via one and the other pathway, respectively (Figure 1, Mechanisms #4). This consideration of basic pyrimidine biology suggests that the combination or sequencing of these 2 drugs could be worth evaluating.
A critical challenge is to promote recovery by functionally normal stem cells that have been depleted by age and other insults and are needed to relieve cytopenia after suppression of malignant clones (Mechanisms #2). Thoughtful combination therapy with thrombopoietin analogs could be one way to address this problem.45-47
Other drugs
The experience with decitabine and 5-azacytidine demonstrates the clinical safety and benefits of using p53-independent pathways and a transcription factor difference between aggressive clonal cells and normal hematopoietic stem cells to terminate malignant growth (Mechanisms #1). Additional, noncytotoxic, chromatin-relaxing drugs to further build this exciting treatment paradigm will no doubt become available for clinical trials. Some kinase inhibitors (eg, potent FLT3 inhibitors) also seem to terminate myeloid clone growth by noncytotoxic mechanisms (possibly by activating proliferation-terminating differentiation genes and/or directly affecting MYC protein stability55 ).
Conclusion
Except in older females, the survival time for a diagnosis of MDS in the United States does not seem to have substantially improved since the introduction of 5-azacytidine into the market in 2004 (Figure 3). However, clinical and mechanism data suggest that judicious, mechanism-based application and optimization of these agents will have increasingly meaningful impact. Moreover, these drugs are exemplars of an inspiring alternative to the traditional cytotoxic paradigm and should pave the way for additional relatively nontoxic but broadly effective agents.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: alternative dosages, schedules, and routes of administration of decitabine, a drug approved by the FDA for the indication of MDS.
Correspondence
Yogen Saunthararajah, MD, Lerner College of Medicine of Case Western Reserve University, 9500 Euclid Avenue, R40, Cleveland, OH 44195; Phone: 216-444-8170; Fax: 216-636-2498; e-mail: saunthy@ccf.org.
