
Abstract
Sickle cell disease (SCD) is the most common inherited hemoglobinopathy. Despite antenatal counseling and neonatal screening programs implemented in higher income countries, SCD is still associated with multiple morbidities and early mortality. To date, the only curative approach to SCD is hematopoietic stem cell transplantation, but this therapy is not yet established worldwide. The registries of the European Blood and Marrow Transplant (EBMT) and the Centre for International Blood and Marrow Transplant Research (CIBMTR) account, respectively, for 611 and 627 patients receiving transplantations for SCD. Most of these patients were transplanted with grafts from an HLA-identical sibling donor. The main obstacles to increasing the number of transplantations are a lack of awareness on the part of physicians and families, the absence of reliable prognostic factors for severity, and the perceived risk that transplantation complications may outweigh the benefits of early transplantation. Results show that more than 90% of patients having undergone an HLA-identical sibling transplantation after myeloablative conditioning are cured, with very limited complications. Major improvement is expected from the use of new reduced-toxicity conditioning regimens and the use of alternative donors, including unrelated cord blood transplantations and related haploidentical bone marrow or peripheral blood stem cell transplantations.
Introduction
Sickle cell disease (SCD) is a severe hereditary hemoglobin disorder. In the absence of appropriate long-term management, it leads to death in early childhood or to later organ failure (brain, skeleton, heart, lungs, or kidneys). It mainly affects countries where malaria is or has been highly prevalent. SCD predominates in Africa and is also highly prevalent in India, Brazil, and the Caribbean Islands, but, due to the flow of immigration, the disease has now also spread to the rest of the world. In the 1970s, 10% of American SCD patients died before their 10th birthday and 50% before their 21st birthday; today, the average life expectancy is 40 to 50 years.1 Despite progress made in management of this disease, such as the prevention of pneumococcal infections, the introduction of hydroxycarbamide, and the early detection of cerebral vasculopathy with transcranial Doppler, SCD remains a systemic disease with a high risk of morbidity and mortality. Most SCD patients live in low-income countries where basic standard care is not easily accessible. In the United States, Brazil, and most European countries, neonatal screening has proven to be an invaluable tool for the control of SCD. Unfortunately, neonatal screening is not implemented worldwide and some patients are diagnosed only when they present with complications. Diagnosis at birth followed by early prophylactic treatment with penicillin and vaccination programs against common pathogens can significantly reduce the rate of early death from infections. However, there are still many low-income countries where the facilities for even simple screening remain unavailable. Worldwide, more than 350 000 children are born each year with a severe inherited hemoglobinopathy and 80% are born in low- or middle-income countries. In Africa, the number of newborn children with SCD is estimated to be 200 000 to 300 000. In developed countries, SCD is a disease of increasing public health concern. In France, the current prevalence is ∼ 12 000 to 15 000 patients, with 400 newborns diagnosed every year. In the United States, 70 000 to 100 000 patients live with SCD.
There is presently no curative therapy for SCD other than HLA-identical sibling allogeneic hematopoietic stem cell transplantation (HSCT).2,3 However, despite a cure rate of > 90%, HSCT is not available to most patients because of their socioeconomic setting or because of the absence of a matched related donor. The risks of transplantation-related mortality (TRM), GVHD, and infertility have a negative effect on the referral rates from primary care physicians to transplantation centers. Research is ongoing to increase the HSCT donor pool, including mismatched cord blood (CB) and haploidentical related donors, and to decrease toxicity related to conditioning regimens.
Indications for HSCT in patients with homozygous SCD
Today, due to progress in diagnostics and follow-up, 95% of SCD patients are alive at 15 years of age. Furthermore, hydroxycarbamide, transfusions, and iron chelation have made it possible to address certain SCD complications and most patients now reach adulthood.4-6 Although survival has improved significantly in the last 2 decades, the mortality rate continues to increase dramatically once patients reach adulthood. SCD-associated morbidity and mortality in young adults is largely due to currently unpreventable complications such as priapism, avascular osteonecrosis, chronic pulmonary impairment, hypertension, stroke, sickle nephropathy, and recurrent venoocclusive crises. In addition, transition in care from the pediatric to the adult medical setting often results in loss of follow-up and increased risk of morbidity and mortality.7
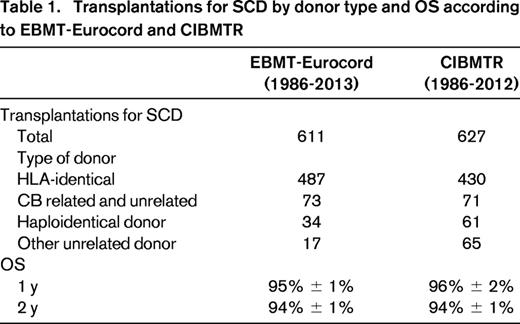
Prognostic criteria of severity for SCD are debated. In the absence of robust epidemiological studies, the percentage of SCD patients requiring transplantation remains unknown; however, it has been estimated at 10% to 50% in children and increases with age. In the United States, the estimated number of homozygous SCD patients is 70 000 to 100 000, of which ∼ 5 000 to 7 000 could be eligible for transplantation. Despite this, the total number of transplanted patients reported in the Centre for International Blood and Marrow Transplant Research (CIBMTR) is ∼ 600 and the same scenario is observed in Europe. This demonstrates that HSCT for SCD represents an unmet need, even in developed countries (Table 1).
There is no general agreement on referral of patients with SCD to an HSCT program. The decision can depend on factors including patient age, existence of severe comorbidities, HLA-identical donor availability, and sociocultural setting. Indications for transplantation are broader and less stringent in patients with genoidentical sibling donors, for whom transplantation could be considered the standard of care. The decision to provide transplantation is difficult because of the lack of good indicators of severity (Table 2). These criteria may change according to the patient's age and the type of donor and with the implementation of new conditioning regimens.

Results of HSCT in SCD
A recent survey of the European Blood and Marrow Transplant (EBMT) and CIBMTR data files shows that ∼ 1200 patients in total have received transplantations in Europe and in the United States. Most received an HLA-identical sibling transplantation (Table 1). The 3-year overall survival (OS) is ∼ 90% regardless of the source of HSCs (Figure 1).

OS according to donor source. (A) OS in the United States (CIBMTR data). (B) OS in Europe (EBMT data). CIBMTR data show that the 1-year probability of OS for SCD patients for HLA-identical = 91%, CB = 84%, unrelated donor = 71%, and haploidentical = 91%; EBMT data show that the 1-year probability for OS for SCD patients for HLA-identical = 95%, CB = 96%, unrelated donor chest = 92%, and haploidentical = 78%.
OS according to donor source. (A) OS in the United States (CIBMTR data). (B) OS in Europe (EBMT data). CIBMTR data show that the 1-year probability of OS for SCD patients for HLA-identical = 91%, CB = 84%, unrelated donor = 71%, and haploidentical = 91%; EBMT data show that the 1-year probability for OS for SCD patients for HLA-identical = 95%, CB = 96%, unrelated donor chest = 92%, and haploidentical = 78%.
HLA-identical sibling HSCT
Myeloablative conditioning
HLA-identical sibling donors represent the majority of transplantations for SCD worldwide (Table 3). Most centers use a myeloablative conditioning regimen consisting of the combination of busulfan (BU), cyclophosphamide (CY), and antithymocyte globulin (ATG) or alemtuzumab. In the largest series by Bernaudin et al, OS and event-free survival were 93% and 86%, respectively. A major finding was that the rejection rate dropped from 22.6% to 3% when ATG was added to the conditioning regimen.3 The results of several major studies summarized in Table 3 show an OS of > 90% and an event-free survival of > 80%.3,8-10 After encouraging results were obtained through related CB transplantation (CBT) for hemoglobinopathies,11 Eurocord performed a study comparing the outcome of patients with hemoglobinopathies given either CBT or BM transplantation (BMT) from an HLA-identical sibling. Among the SCD patients, 130 received BMT and 30 received CBT. Compared with the patients who received BMT, those having received CBT had slower neutrophil recovery and less GVHD. None of the CBT patients developed extensive chronic GVHD. The 6-year disease-free survival was 92% after BMT and 90% after CBT.12 Therefore, CB from an HLA-identical sibling appears to be a suitable source of stem cells for HSCT in SCD providing that the CB units collected and cryopreserved at birth comprise > 3.5 × 107/kg total nucleated cells (TNCs). The use of related CB spares the young donor a BM aspiration under anesthesia and decreases the probability of acute and chronic GVHD in the recipient. CB cells from HLA-identical siblings present further advantages, in particular in African countries, because the cells are collected before the donor is likely to contract an infectious disease and before he or she can become lost to follow-up.
Summary of the largest studies on HLA-identical HSCT in SCD
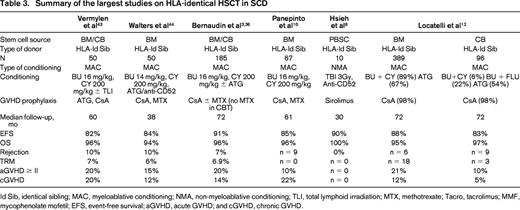
Id Sib, identical sibling; MAC, myeloablative conditioning; NMA, non-myeloablative conditioning; TLI, total lymphoid irradiation; MTX, methotrexate; Tacro, tacrolimus; MMF, mycophenolate mofetil; EFS, event-free survival; aGVHD, acute GVHD; and cGVHD, chronic GVHD.
The cost of collecting cells at birth from healthy donors in families with a history of SCD should be calculated. If the CB of an HLA-identical sibling donor is immediately available, the transplantation can be scheduled early, before the patient experiences any major complications. Unfortunately, related CBTs are not often performed because most patients do not have a pregnant mother and there is a limited number of directed CB banks for family use.13 Preimplantation genetic diagnosis and embryo selection have been successfully performed in some cases to select a healthy HLA-identical sibling for CBT.14,15
Overall, the short- and long-term results of HLA-identical sibling HSCT are excellent with very little TRM.16-18 Long-term result analysis performed by Bernaudin et al showed a long-term hematological reconstitution with full donor chimerism.3 Quality of life was markedly improved. In the majority of cases, it was possible to interrupt immunosuppression between 6 and 9 months after HSCT and the function of most organs was improved. There was no recurrence of venoocclusive crises, stroke, or acute coronary syndrome. Most patients from Africa were able to return home after 1 year and, in absence of GVHD or splenectomy, prophylactic penicillin was stopped 2 years after transplantation. Most patients resumed normal school activity and improved their learning performances. Growth and development varied according to sex and age at transplantation and some patients needed hormone replacement.
Sterility is a major concern after myeloablative conditioning. Pretransplantation sperm and ovarian cryopreservation is highly recommended. Some recent studies have reported successful pregnancies after ovarian preservation and transplantation19-22 and continuing advances in fertility medicine will hopefully further benefit HSCT patients.
These results indicate that HLA-identical sibling HSCT after myeloablative conditioning with ATG should be considered as the standard of care for SCD children. In view of the probability that 95% of SCD children will be cured after HLA-identical sibling HSCT, this therapeutic approach should be discussed with the family early on, as soon as long-term transfusions become necessary or when signs of latent infarction (defined as ischemic lesions on MRI without clinical neurological symptoms) are detected.
Reduced-intensity conditioning regimens
All of these excellent results were obtained through the use of a myeloablative conditioning regimen, including some encouraging results in adults. However, short- and long-term toxicity is a concern and some researchers have attempted to use reduced-intensity conditioning (RIC) with an HLA-identical donor for adults with SCD. The first attempts were not very successful, with a high rate of non-engraftment. Better results have been reported more recently after modification of the conditioning regimen. Hsieh reported a phase 1/2 trial in 10 adult patients who received conditioning with alemtuzumab and 3 Gy of total body irradiation (TBI), followed by an HLA-identical related peripheral blood stem cell (PBSC) transplantation. GVHD prevention consisted of sirolimus. Eight of the 10 patients engrafted and were alive with a median follow-up of 30 months and there was a high incidence of mixed chimerism.8 In other smaller series, patients received various conditioning regimens including low-dose busulfan (BU) or total body irradiation (TBI) with fludarabine (FLU) anti-thymocyte globulin (ATG)/alemtuzumab or melphalan. In most cases, the conditioning was well tolerated with > 90% OS, but with a high rate of mixed chimerism, rejection, and autologous reconstitution.23-25 There is a growing need to reach a common consensus protocol to determine which regimens reach the goal of full or mixed chimerism and limited short- and long-term toxicity.26 Other drugs are under investigation for conditioning in SCD patients. In other settings, treosulfan was used instead of BU because it is well tolerated. Thiotepa has been shown to increase engraftment in CBT.25,27
Despite the encouraging results of HSCT with an HLA-identical sibling, the number of SCD patients receiving transplantations remains low. Systematic HLA typing is still not performed in most families. In some countries, the cost of HLA typing is covered by public or private insurance programs and this contributes to increased access to HSCT.
Walters et al reported that, among 4848 SCD patients younger than 16 years, 315 (6.5%) met the criteria for transplantation. However, only 128 (41% of the eligible or 2.6% of the total) underwent HLA typing. Reasons for not performing HLA typing were lack of sibling donor (24%), lack of financial or psychosocial support (10.5%), parental refusal (9.5%), and physician refusal (4%).9 In view of the good results and reduced toxicity obtained in recent years with HSCT in SCD patients, campaigns to educate health care providers and family support groups about HSCT options for SCD are warranted.
Alternative donor transplantations
Probability of finding an alternative donor for HSCT in SCD
The probability of finding a suitable donor has increased over the years (www.BMDW.org). There are currently more than 21 million unrelated donors listed in BM donor registries worldwide, including 20 971 716 BM and peripheral blood adult volunteer donors and 571 318 CB units. Despite this large number of registered potential donors worldwide, registries still find it difficult to identify donors from ethnic minorities.28,29 A recent National Marrow Donor Program (NMDP) investigation found that whereas ∼ 79% of whites searching the NMDP registry found at least one 8/8 HLA-allele matched potential donor, only 50% of Asian/Pacific islanders, 44% of Hispanics, and 33% of African Americans found a similarly HLA-compatible potential donor (personal communication). When looking at the current NMDP inventory, which contains approximately 150 000 CB units, the probability of finding an 8/8 allele-matched donor in African-American patients is 5%, a 7/8 match 33%, and a 6/8 mismatch 80% (M. Eapen, unpublished results, June 2013). This probability may be higher at the level of the world inventory and would increase further through improved knowledge of the most frequent haplotypes found in African populations.
It is difficult to analyze the difficulties that currently hinder the search for a donor in the setting of ethnic minorities. Africans have less common and more diverse haplotypes than the white population. Furthermore, multiple culturally related and psychosocial barriers can lead to ambivalence about donation. In our own experience, African families are more willing to donate CB for a family member or for someone else than they are to donate adult BM or PBSCs.
Results of alternative donor transplantations
There are very few reports of matched unrelated donor transplantation for patients with SCD because ethnic variability is underrepresented in the registries,29 making it difficult to find a suitable donor. For this reason, most HLA-mismatched transplantations are from unrelated CB or related haploidentical donors.
Unrelated CBTs
Eurocord has reported the outcome of unrelated mismatched CBTs in 16 patients with SCD.30 Ten patients received myeloablative conditioning with either BU-CY (n = 7) or BU-FLU (n = 3) and 6 patients received a RIC regimen containing BU-FLU (n = 3), melphalan-FLU (n = 2), or CY-FLU (n = 1). Fourteen of these patients received ATG or alemtuzumab. At 2 years, 94% were alive and 53% were disease free. Engraftment was associated with the number of total nucleated cells (TNCs) in the CB unit. Engraftment for patients who received a median > 5 × 107 TNCs/kg was 68% at a median time of 16 days compared with 30% for patients who received < 5 × 107 TNCs/kg. This series was too small to analyze other factors such as age, HLA compatibility, conditioning, and transplantation year. Another study reported similar results with a conditioning regimen associating BU-FLU and alemtuzumab.31 Kamani et al reported a phase 2 trial in 8 patients transplanted with CB and conditioned with alemtuzumab-FLU-melphalan.32 All of the patients recovered blood counts in a median time of 22 days; 5 had autologous reconstitution and 3 full donor chimerism; and 2 patients had grade II GVHD. Overall, 7 patients were alive and 3 disease free. Based upon the high incidence of graft rejection, enrollment in the CB arm of the trial was suspended.
Haploidentical related donors
Bolanos et al reported a phase 1/2 trial on adult patients with SCD who received a haploidentical related BMT (some patients received G-CSF–primed BM).33,34 The conditioning consisted of ATG (days −9 to −8), CY 14.5 mg/kg × 2 (days −6 to −5), FLU 30 mg/m2 × 5 (days −6 to −2), and 2 Gy of TBI (day −1). After transplantation, patients received CY 50 mg/kg on days +3 and +4, followed by GVHD prevention with mycophenolate mofetil and tacrolimus or sirolimus. All patients recovered blood counts at a median time of 24 days. Graft failure was observed in 43% of patients. Eleven patients had evidence of long-term engraftment and 6 of them now have full myeloid chimerism and are off of immunosuppression. Mixed chimerism ranged from 49% to 63% CD3+ cells. No GVHD was observed. On short-term follow-up, all patients were alive and 11 of 17 were disease free. Despite the high number of rejections, these results are encouraging because of the low toxicity of the procedure. It is possible to increase the probability of finding a donor by taking haploidentical related donors into consideration, because the majority of patients have a suitable parent, child, or haploidentical sibling.
These promising data need to be confirmed in a larger cohort of patients and compared with other HSC sources (eg, G-CSF–mobilized BM or PBSCs), conditioning regimens, and GVHD prevention.35,36 There are numerous phase 1/2 studies listed at www.clinicaltrials.gov.
Future directions and conclusion
HSCT for patients with SCD, although highly effective, is underused. The cumulative data on results of myeloablative HSCT in children indicate that HSCT from an HLA-identical sibling should be performed early in the course of the disease. Cost-benefit and quality-of-life analyses should be performed to compare these strategies. Using data from individuals diagnosed with SCD enrolled in the Florida Medicaid program between 2001 and 2005, Kauf et al calculated that the lifetime cost of care averages $460.15 per patient.37 This is comparable to the cost of HSCT from sibling donors for nonmalignant diseases, which ranges from $112 000 to $150 000 ($1.900 per expected life year) when HSCT is performed during childhood.
The ultimate objective in the battle against SCD is to systematically perform family HLA typing early in life and to install primary care centers able to detect early complications. In addition, it is important to design and investigate new RIC regimens able to decrease HSCT complications and to preserve fertility. This would also decrease the costs of the transplantation process and make transplantation more easily accessible in developing countries, where cost is a major obstacle to generalized global treatment of SCD.
The use of alternative donors is still under investigation. The choice between an unrelated CB and a related haploidentical donor will depend on the results of studies comparing these 2 procedures. Randomized studies and registry-based comparative studies are ongoing and will provide the basis for recommendations for donor selection (Figure 2).

It should be possible to make HSCT more accessible to a large majority of patients by improving our understanding of how to overcome the limitations of alternative donor transplantation, including delayed engraftment and poor immune cell reconstitution. Our current experience with CBT for nonmalignant disease shows that the best results are obtained using a 6/6 or 5/6 HLA-matched CB, provided that the TNC content is > 5 × 107/kg.38 In addition, patients who have anti-HLA antibodies against the CB unit should not receive this cord blood unit.39 Other factors should also be taken into consideration, including ABO compatibility, high-resolution HLA typing, and noninherited maternal antigen mismatches. If the number of cells is insufficient in a single CB unit, infusion of 2 units could be indicated. Results in SCD are not known, but in other malignant diseases, double CBT gives better engraftment and more GVHD.38
To improve CBT results, several options are being explored. Intrabone injection to enhance the homing process known to be highly inefficient after IV cell delivery has been attempted. In a European study, intrabone injection had a significant advantage in terms of engraftment and decreased GVHD.40
Many investigators have explored CB expansion strategies as a way to increase cell dose. Using the notch ligand Delta 1, Delaney et al demonstrated expansion of short-term repopulating cells and an improvement in the time of neutrophil engraftment to 16 days.41 The M.D. Anderson group used a coculture ex vivo with mesenchymal progenitor cells in 1 of 2 CB units given to 31 patients and reported a 30-fold expansion in CD34+ count and a median time to engraftment of 15 days.42 Related haploidentical transplantations using various technologies and stem cell sources with either T-cell–depleted or unmanipulated T-cell–replete G-CSF–primed BM or PBSCs have been used, mostly in patients with hematological malignancies, with a high rate of engraftment and low TRM. The use of CY after transplantation was pioneered by the Baltimore group, with a low incidence of acute and chronic GVHD. Although the results are encouraging, more patient accrual and longer follow-up times are needed to strengthen their results.
In conclusion, HSCT currently remains the only available treatment to cure hemoglobinopathies. The results obtained in several studies seem to be in favor of early transplantation if a suitable donor is available. Current transplantation outcomes have improved considerably compared with those from the 1980s and 1990s. Nowadays, more than 90% of patients survive transplantation and more than 80% of them become disease free. This pertains to patients treated in centers worldwide, including those in developing countries.
Disclosures
Conflict-of-interest disclosure: The author has received honoraria from Cord Use and Gamida. Off-label drug use: None disclosed.
Correspondence
Pr E. Gluckman, Eurocord, Hospital Saint Louis, 1 avenue Claude Vellefaux, 75010 Paris, France; Phone: 33609489715; Fax: +33142492699; e-mail: eliane.gluckman@sls.aphp.fr.
