
Abstract
Characterization of the molecular basis of the β-thalassemias and sickle cell disease (SCD) clearly showed that individuals with the same β-globin genotypes can have extremely diverse clinical severity. Two key modifiers, an innate ability to produce fetal hemoglobin and coinheritance of α-thalassemia, both derived from family and population studies, affect the pathophysiology of both disorders at the primary level. In the past 2 decades, scientific research had applied genetic approaches to identify additional genetic modifiers. The review summarizes recent genetic studies and key genetic modifiers identified and traces the story of fetal hemoglobin genetics, which has led to an emerging network of globin gene regulation. The discoveries have provided insights on new targets for therapeutic intervention and raise possibilities of developing fetal hemoglobin predictive diagnostics for predicting disease severity in the newborn and for integration into prenatal diagnosis to better inform genetic counseling.
Introduction
Among the hemoglobinopathies, sickle cell disease (SCD) and β-thalassemia have the most impact on morbidity and mortality, affecting millions worldwide.1 Both are prototypical Mendelian single gene disorders affecting the β-globin (HBB) gene. Despite the apparent genetic simplicity, both disorders display extreme clinical heterogeneity.2,3 Major advances have been made in our understanding of the molecular pathology, pathophysiology, and treatment of these disorders, but predicting the clinical course remains difficult. This is particularly so for SCD, which presents challenges in genetic counseling and management. Identification of the genetic modifiers could provide more precise estimates of disease severity and defining the genetic variation within the pathobiological pathways could provide clues and targets for therapeutic intervention.
The 2 major modifiers, an innate ability to produce fetal hemoglobin (HbF, α2γ2) and coinheritance of α-thalassemia, have been derived from more than 50 years of extensive biochemical and pathophysiological studies and were subsequently confirmed by genetic studies. The mechanisms of the modifying effects of HbF and α-thalassemia in SCD and β-thalassemia could not be more different at the molecular level,yet these genetic modifiers have a large clinical effect due to their impact on disease pathophysiology at the primary level. In addition, the genetic variants are common and their contribution to disease burden is substantial.
Rationale for genetic studies and approaches used to demonstrate genetic modifiers
HbF levels and coinheritance of α-thalassemia are unlikely to be the only major modifiers because they do not completely explain the clinical diversity in either disorder. In SCD, for example, analysis of sibling pairs suggested a genetic contribution to stroke risk not explained by HbF levels or coinheritance of α-thalassemia; a child with sickle cell anemia (SCA) has an increased risk of stroke if they have had siblings who have experienced an overt stroke. Studies have also shown a concordance in response to hydroxyurea therapy among siblings. Observations of clinical variability between identical twins (who have identical genetic make-up)4 and longitudinally within the same individual highlight the important contribution of environmental factors to the phenotypic variability. Environmental factors such as physical activity, diet, and toxins can elicit changes in the gene activity (altering epigenome or “software”) without changing the DNA code (or “hard-drive”). Such epigenetic changes may account for the discordant clinical phenotypes in identical twins and contribute to the complex disease etiology in both disorders.5 Due to the complex interaction of the multiple factors, a genetic approach might be the most efficacious way of identifying the modifier genes and provide a better understanding of the disease mechanisms.
All 3 approaches that are generally used to unravel genetic modifiers in human disorders have been applied to the β-hemoglobinopathies: mouse models, family and epidemiological studies, and genetic association studies. Although mouse models of β-thalassemia and SCD2 have been very important in providing proof-of-principle for globin gene regulation along with development and testing of therapeutic compounds, no novel genetic modifiers have been discovered using this approach. In part, this may be due to a lack of mouse models that replicate the complications encountered in SCD (eg, stroke). Insights into the 2 major genetic modifiers, HbF levels and α-globin genotype, were derived from an understanding of the disease pathophysiology and were subsequently validated by family and population genetic association studies (Tables 1, 2). Association of serum bilirubin levels and predisposition to gallstones with polymorphisms in the promoter of the UGT1A1 gene (Gilbert syndrome) have been consistently demonstrated in many studies in patients with β-thalassemia and SCD of all ages and different populations.6,7 A recent single study suggested that the MYH9-APOL1 locus, an important genetic risk factor for renal failure in non-SCD population of African ancestry, is also associated with sickle cell nephropathy.8 These association studies were based on candidate genes selected according to one's understanding of the disease pathophysiology or prompted by association with the same phenotypes in non-SCD populations.9
Genetic modifiers of SCD

Most modifiers that affect SCD at the secondary level of subphenotypes and complications have not been replicated.
Candidate gene association studies look for differences in the frequencies of genetic variants in targeted genes between cases and controls. If a variant is more common in cases than controls, then association can be inferred. The more recent genome-wide genetic association studies (GWAS) involve an unbiased scan of the whole human genome and, by design, are more likely to reveal unsuspected interactions. GWAS will also confirm previous candidate genes if the association is robust.10,11 A case in point is the application of GWAS in the highly successful discovery of BCL11A (an oncogene that hitherto was not known to have a role in erythropoiesis) as a quantitative trait locus (QTL) controlling HbF.10,12 GWAS also confirmed association of the other 2 loci, Xmn1-HBG2 (rs782144) on chromosome 11p and HBS1L-MYB (HMIP) on chromosome 6q, which were previously discovered through candidate and genetic linkage studies, with HbF production. Similarly, GWAS confirmed the association between bilirubin level and UGT1A1 polymorphism in SCD.11
It has become clear from the genetic association studies of HbF and other common diseases and traits that GWAS can work13 but that sample size matters, that clearly defined and well harmonized phenotypes are critical, that replication and collaboration (interdisciplinary in addition to increasing sample size) matters, that current hypotheses regarding candidate genes and pathways may not matter so much, and that several genes can influence more than one disease or trait. It has also become evident that simpler phenotypes such as HbF, which are reproducible, measurable, and disease related, are much more robust and successful in genetic association studies than clinical end points. Such intermediate end points or endophenotypes are often quantitative traits and thus provide more power in genetic strategies. For quantitative continuous traits, one could focus on extremes of the trait as a strategy to reduce genotyping or sequencing costs, as was successfully applied in the GWAS for HbF and F cells.10
The brain is one major site of morbidity in children with SCD. Increased velocity in the middle cerebral artery as detected by transcranial Doppler (TCD) screening is a biomarker of early cerebrovascular disease. Studies have shown that chronic blood transfusion therapy at this stage can prevent overt stroke. True primary stroke prevention, however, should prevent vascular damage before TCD velocity becomes abnormal. TCD velocity would therefore be an extremely attractive endophenotype in studies for detecting genetic variants associated with sickle vasculopathy and stroke risks.
Whole genome or exome sequencing using next-generation sequencing technology in combination with well-defined phenotypes offers the possibility of identifying new genetic variants. GWAS in combination with exome sequencing identified mutations in GOLGB1 and ENPP1 with stroke protection in SCA.14 In this study, overt stroke was the clinical marker, but these variants have yet to be independently validated in a different population group.
Review of genetic modifiers in β-hemoglobinopathies
For both SCD and β-thalassemia, factors that affect the primary event of the disease process will have a global effect on the disease phenotype. These include the causative genotype, coexisting α-thalassemia, and the innate ability to produce HbF.
SCD should be considered as both a qualitative and quantitative genetic disorder in that it is caused by the presence of an abnormal Hb variant (HbS, α2β2S, HBB glu6val, GAG β6 GTG), yet the likelihood of HbS polymerization and sickling is highly dependent on the intra-erythrocyte HbS concentration. Homozygosity for HbS (SCD-SS) or SCA is the most common genotype; the other causative genotypes include compound heterozygous states of HbS with HbC (SCD-SC) or β-thalassemia variants (SCD-Sβ0 thalassemia and SCD Sβ+ thalassemia).2 Individuals with SCD-SS or SCD-Sβ0 thalassemia, in whom the intracellular hemoglobin composition is almost all HbS, have the most severe disease, followed by SCD-SC and SCD Sβ+ thalassemia. Simple heterozygotes for HbS (HbAS) who have 30% to 40% intracellular HbS, with HbA in excess of HbS, are asymptomatic. Under exceptional circumstances, however, such as intense physical activity and dehydration, the consequent increased intracellular HbS concentration can induce vasoocclusive pain.
Approximately one-third of SCD patients of African descent have coexisting α-thalassemia due to the common deletional variant (−α3.7/).2 The majority are heterozygous (αα/α−), with 3% to 5% homozygous for the deletion (α−/α−). Coexisting α-thalassemia reduces intracellular hemoglobin concentration, thereby reducing HbS polymerization, reducing sickling, and decreasing hemolysis. Although the coexisting α-thalassemias have a protective effect against complications associated with severe hemolysis, such as priapism, leg ulceration, and albuminuria, the increased hematocrit and blood viscosity may account for the increase in other complications associated with microvascular occlusion, such as increased acute pain, acute chest syndrome, osteonecrosis, and retinopathy.15 Coexisting α-thalassemia also reduces bilirubin with a quantitative effect that is independent to that of the UGT1A1 promoter polymorphism.7 In Jamaicans, the absence of α-thalassemia and higher HbF levels predict a benign disease. A subsequent analysis of 40 elderly Jamaican patients (> 60 years of age), however, suggested that coexisting α-thalassemia may promote longevity (51% had coexisting α-thalassemia), but the trend failed to reach significance.16 Coinheritance of α-thalassemia blunts the response to hydroxyurea therapy in SCD, which may be explained by its effect on HbF levels and mean cell volume, 2 key parameters associated with hydroxyurea response.17 It is quite likely that α-thalassemia carriers could have a poorer response to RBC membrane channel blockers that aims to reduce sickling through preservation of cell hydration.
HbF reduces the propensity for HbS polymerization; the hybrid tetramers (α2β2γ) inhibit HbS polymerization and the presence of HbF dilutes down the intracellular HbS concentration. In view of its impact at the primary level of disease pathology, one would expect HbF levels to have a global beneficial effect. Indeed, HbF levels are a major predictor of survival in SCD, and low levels of HbF have been associated with increased risk of brain infarcts in young children.18 At the subphenotype level, apart from the clear benefit of high HbF levels with acute pain and leg ulceration, there are disparities and less conclusive evidence for its effects on other complications such as stroke, renal impairment, retinopathy, and priapism.15 These disparate conclusions are likely to arise from small sample sizes, even smaller numbers of end complications, and disparities in ascertainment of phenotypes. The uneven beneficial effect of HbF on sickle-related complications could also be related to the different pathobiology of large and small vascular disease.
HbF levels vary considerably, from 1% to as high as 25% in individuals with SCD-SS, and behave as a quantitative genetic trait as in healthy individuals. Three QTLs, one in cis to the HBB gene cluster represented by the Xmn1-HBG2 site (rs7482144), HBS1L-MYB intergenic polymorphisms (HMIP) on chromosome 6q, and BCL11A on chromosome 2p, are major regulators of common HbF variation.10,12,19-22 Their effect on HbF levels varies with the frequency of the HbF boosting (minor) alleles in different population groups. In patients of African descent with SCD, the 3 loci account for 16% to 20% of the variation in HbF levels with a corresponding reduction in acute pain rate.23,24 The favorable effect of HbF and its modifiers (BCL11A and Xmn1-HBG2) variants on pain and TCD velocities is measurable even in young infants, as demonstrated in the recently completed BABY HUG study.25
Several studies have investigated the association of candidate genes implicated in pathophysiology of vasoocclusion and vasculopathy, such as those encoding factors modifying inflammation, oxidant injury, nitric oxide biology, vasoregulation, and cell adhesion, with sickle-related complications, including stroke, priapism, leg ulcers, avascular necrosis, renal disease, acute chest syndrome, gallstones, and susceptibility to infection.15,26,27 The majority of the reported associations have not been replicated or validated and are likely to be false positives. Of the numerous association studies reported, the most robust is the association between serum bilirubin levels and predisposition to gallstones with the 6/7 or 7/7 (TA) repeats in the UGT1A1 promoter.11 UGT1A1 polymorphism continued to have a strong influence even during hydroxyurea therapy; in one study, children with the 6/6 UGT1A1 genotype achieved normal bilirubin levels, whereas children with 6/7 or 7/7 UGT1A1 genotypes did not.6
Suggestions of a familial predisposition to stroke and its devastating consequences has prompted numerous genetic and clinical association studies on cerebrovascular complications involving either large or small vessels.14,28,29 Of the 38 published single nucleotide polymorphisms (SNPs) associated with stroke, the effects of α-thalassemia and SNPs in four genes (ADYC9, ANXA2, TEK, and TGFBR3) could be replicated, although only nominally significant association results were obtained.30 More recently, GWAS in combination with whole-exome sequencing have identified mutations in 2 genes, GOLGB1 and ENPP1, which are associated with reduced stroke risk in pediatric patients, but, again, this needs validation in independent studies.14 To overcome the small sample size in end point complications, a study used a compound phenotype that included one or more sickle-related complications (see review by Thein27 ). Patients with complications had a higher frequency of the platelet glycoprotein allele HPA-5B. In this small study, most of the complications were osteonecrosis and only 4 individuals had more than one complication. Because traditional methods are often inadequate in association studies of complex traits, methods of evaluating multilocus data are promising alternatives. A GWAS was applied to SCD based on a disease severity score that was derived from a Bayesian network that integrates 25 different clinical and laboratory variables.31 Several genes not known to be related to the pathogenesis of SCD were identified, including KCN6 (a potassium channel protein) and TNK5 (a gene encoding tankyrase-1, a possible telomere length regulator). However, it is important to remember that results from such analytical techniques are dependent on the structure of a model assuming certain causalities and probabilities of the different variables. More recently, GWAS identified an SNP (rs7203560) in NPRL3 on chromosome 16p that was independently associated with hemolysis (using a score derived by principle component analysis).32 rs7203560 is in perfect linkage disequilibrium with SNPs within the α-globin gene regulatory elements (HS-48, HS-40, and HS-33). rs720356 is also in linkage disequilibrium with ITFG3 that is associated with RBC mean cell volume and mean cell hemoglobin in several GWAS of different population groups.33 It is proposed that NPRL3 reduces hemolysis through an independent thalassemic effect on the HBA1/HBA2 genes.
Hydroxyurea remains a major treatment option for SCD; its main effect is mediated primarily through induction of HbF. Clinical response to hydroxyurea therapy, however, is variable with variable HbF response; a main determinant of response appears to be the baseline HbF levels. Numerous genetic association studies on HbF response to hydroxyurea have been reported, of which the association with Xmn1-HBG2 seems to be the most robust. In the recently completed BABY HUG trial, however, the HbF genetic modifiers were not able to identify the “high” and “low” responders to hydroxyurea.25 Table 1 summarizes the genetic modifiers of SCD.
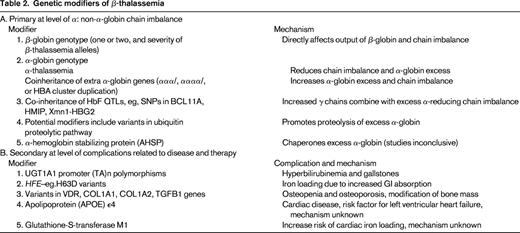
In β-thalassemia, as in SCD, the causative genotype, coinheritance of α-thalassemia and HbF, are the main modifiers of clinical severity. These genetic factors have a major impact because they affect the central mechanism underlying disease pathophysiology (ie, the degree of globin chain imbalance and the excess of α-globin chains). Almost 300 mutations (deletions and point mutations) that down-regulate the β-gene have been described (http://globin.bx.psu.edu/hbvar/menu.html). Functionally, the mutations range from null mutations (β0 thalassemia) that cause a complete absence of β-globin production to those that cause a minimal deficit: β++ thalassemia (sometimes referred to as silent β-thalassemia because of the minimally reduced RBC indices and normal HbA2 levels in heterozygotes).34 The causative β-globin genotype has the largest impact on disease severity. Homozygotes or compound heterozygotes for 2 β0 thalassemia alleles cannot produce any HbA (α2β2); such individuals generally have the most severe anemia and depend on blood transfusions for life (thalassemia major).
In many populations in which β-thalassemia is prevalent, α-thalassemia also occurs at a high frequency. Individuals who have coinherited α-thalassemia have less redundant α-globin and tend to have less severe anemia. The degree of amelioration depends on the severity of the β-thalassemia alleles and the number of functional α-globin genes. At one extreme, patients who have coinherited the equivalent of only one functioning α-globin (−/α−, HbH genotype) with homozygous β-thalassemia have less severe anemia (thalassemia intermedia) if the β-thalassemia alleles are β+, but a more severe phenotype if they have β0 thalassemia.3
In heterozygous individuals with one β-thalassemia allele, coinheritance of α-thalassemia normalizes the hypochromia and microcytosis, but the elevated HbA2 levels remain unchanged. Increased α-globin production through coinheritance of extra α-globin genes (triplicated, ααα/αα− or ααα/ααα; quadruplicated, αααα/αα; or duplication of the whole α-globin gene cluster, αα/αα/αα) with heterozygous β-thalassemia tips the globin chain imbalance further, converting a typically clinically asymptomatic state to thalassemia intermedia. The severity of anemia depends on the number of extra α-globin genes and the severity of the β-thalassemia alleles.
Increased HbF response as an ameliorating factor becomes most evident in non-transfusion-dependent β0 thalassemia patients who have a mild disease despite the complete absence of HbA.35 In this case, the increased γ-chains combine with the redundant α-globin to form HbF (α2γ2), reducing the chain imbalance. Again, the degree of amelioration depends on the severity of β-thalassemia alleles (β+ or β0) and the number of coinherited HbF genetic determinants (common QTLs including BCL11A, Xmn1-Gγ -158 C→T and SNPs in HMIP on chromosome 6q.12,36-39
At the primary level of chain imbalance, the proteolytic capacity of the erythroid precursors in catabolizing the excess α-globin has often been suggested, but this effect has been difficult to define. α-hemoglobin–stabilizing protein, a molecular chaperone of α-globin, has also been suggested as another genetic modifier, but its impact on disease severity has been inconclusive.40
Genetic variants could also modify the different complications of β-thalassemia that are directly related to the anemia and ineffective erythropoiesis or to therapy, such as iron chelation treatment. The secondary complications include jaundice and predisposition to gallstones, osteopenia, and osteoporosis; iron overload; and cardiac disease (see Table 2 for modifiers of β-thalassemia). The degree of iron loading, bilirubin levels, and bone mass are quantitative genetic traits and thus will be modified by genetic variants regulating expression of these traits.
HbF concentration is a major modifier of disease severity for both β-thalassemia and SCD, prompting decades of study into its regulation in adults.41 The study of HbF quantitative genetics and molecular control of hemoglobin switching mirrors the rapidly evolving focus of genomics research in humans, from the study of natural mutants, gene mapping, and GWAS to applications of the other “-omics” technologies such as comprehensive gene expression profiling and DNA-protein interactions. Studies of the deletions in the HBB cluster associated with heterocellular persistence of fetal hemoglobin (HPFH)2 indicated a “repressive” element in the intergenic region between the Aγ-globin (HBG1) and δ-globin (HBD) genes and enhancer elements downstream of the HBB gene. Point mutations associated with HPFH are clustered in regions of the γ-globin gene promoters that subsequently proved to be binding sites for ubiquitous and erythroid-specific transcription factors. Although the HPFH phenotypes could be reproduced in transgenic mice carrying the human β-globin locus and the mutations shown to alter in vitro binding patterns, unambiguous identification of proteins directly involved in globin switching remained elusive. A variety of transcription factors with roles in globin gene regulation, such as GATA-1, KLF1, and SCL/TAL, were identified, but how they regulate the switch from fetal to adult hemoglobin was still not evident.41
Nonetheless, studies of these HPFH mutants and discovery of the upstream regulatory elements (β-locus control region) triggered a series of experiments and led to the concept that switching of fetal to adult hemoglobin involves 2 mechanisms: autonomous silencing of the fetal globin genes and competitive access of the adult globin gene to the upstream β-locus control region. However, it was difficult to reconcile these concepts of Hb switching with the common variable persistence of HbF in healthy adults and in patients with SCD and β-thalassemia. In many cases, the inheritance patterns of the modestly elevated HbF levels are not clear and not linked to the HBB cluster on chromosome 11. An early example is an extended family of Asian-Indian origin, in which segregation analysis showed that the genetic determinant for HPFH was inherited independently from the HBB cluster and eventually mapped to chromosome 6q23.19 Observations of variable HbF with different βS haplotypes first suggested that the HBB cluster is a prime location for a HbF determinant, represented by the C→T SNP (rs7482144) at position −158 of the Gγ promoter, known as the Xmn1-HBG2 polymorphism.2 By early 2006, developments in genetic tools and genotyping platforms expedited by the International Human Genome HapMap Project enabled the development of GWAS that led to the identification of BCL11A on chromosome 2p16 as another locus modifying γ-globin levels.10,12 The next clue in the hemoglobin-switching puzzle again came from human genetics provided by a Maltese family in which 10 of 27 members had HPFH that segregated independently from the HBB cluster.42 Genetic studies followed by expression profiling of erythroid progenitors identified KLF1 as the γ-globin modifier in this family in which members with HPFH were heterozygous for the nonsense p.K288X mutation in KLF1. Soon, a steady stream of novel mutations in KLF1 associated with increases in HbF were reported in different populations (for review, see Borg et al43 ). The increases in HbF occurred as a primary phenotype or in association with RBC disorders such as congenital dyserythropoietic anemia.
Functional studies in primary human erythroid progenitors and transgenic mice demonstrated that BCL11A acts as a repressor of γ-globin; the silencing effect involves reconfiguration of the HBB locus through interaction with GATA-1 and SOX6 that binds the proximal γ-globin promoters.44,45 Profiling of erythroid cells from the Maltese family showed that KLF1 p.K288X carriers had reduced BCL11A expression.42 KLF1 was also shown to be a direct activator of BCL11A in human erythroid progenitor cells and in transgenic mice.42,46 Previous knockout studies in mice had shown that KLF1 is essential for activation of β-globin expression, but the embryonic and fetal globin genes remain fully activated in the absence of KLF1.47 Collectively, these results have led to the proposal that KLF1 has dual functions in the γ-globin to β-globin switch: KLF1 activates HBB directly and also silences the γ-globin gene indirectly via activation of BCL11A.47
High-resolution genetic mapping and resequencing refined the 6q QTL to variants in a 24-kb region between the HBS1L and MYB gene, referred to as HMIP block 2.19 Work in primary human erythroid progenitor cells48 and transgenic mice showed that this region contains distal enhancers required for MYB activation.49,50 MYB, which encodes the c-MYB transcription factor, is a key regulator of hematopoiesis and erythropoiesis. One suggestion of how MYB modulates HbF levels is via its effects on erythroid proliferation/differentiation balance. A low MYB environment favors accelerated erythropoietic differentiation, leading to the release of early erythroid progenitors that are still synthesizing predominantly HbF.51 Another suggestion is that MYB represses fetal globin gene expression via KLF1 activation of BCL11A.52 A third suggestion is that MYB works via its effects on the TR2/TR4 pathway. MYB up-regulates the nuclear receptors TR2/TR4, which are repressors of the γ-globin gene. Therefore, down-regulation of MYB leads to suppression of the TR2/TR4 pathway and up-regulation of fetal globin gene expression. The emerging network of HbF regulation also includes SOX6; chromatin-modeling factor FOP; the NuRD complex; the orphan nuclear receptors TR2/TR4 (part of direct repeat erythroid definitive [DRED]); the protein arginine methyltransferase PRMT5, involving DNA methylation; and HDACs 1 and 2, which are epigenetic modifiers53 (Figure 1). Regulators of the key TFs, such as microRNA-15a and 16-1, in controlling MYB54,55 could also have a potential role in regulating HbF levels.

Emerging network of γ-globin regulators in adult life and prospective targets for therapeutic induction of HbF. Targets identified in the emerging network of HbF regulation include the KLF1, BCL11A, and MYB genes and the TR2/TR4 nuclear receptors that associate with corepressors DNA methyltransferase 1 (DNMT1) and lysine-specific demethylase 1 (LSD1). KLF1 has a dual role in the silencing of γ-globin genes: it activates BCL11A, a repressor of γ-globin gene expression, and it also activates the β-globin gene directly. BCL11A interacts with the GATA1, FOG1, and SOX6 erythroid transcription factors and with the NuRD deacetylase and remodeling complex to promote suppression of γ-globin gene expression. The nuclear receptors TR2/TR4 associate with corepressors DNMT1 and LSD1 as part of the DRED complex, a known repressor of embryonic and fetal globin genes in adults. MYB contributes to HbF regulation via activation of KLF1 (which activates BCL11A), activation of the DRED complex, and by modulating the number of F cells as part of its effect on erythroid differentiation kinetics and its pleiotropic effect on hematopoiesis.
Emerging network of γ-globin regulators in adult life and prospective targets for therapeutic induction of HbF. Targets identified in the emerging network of HbF regulation include the KLF1, BCL11A, and MYB genes and the TR2/TR4 nuclear receptors that associate with corepressors DNA methyltransferase 1 (DNMT1) and lysine-specific demethylase 1 (LSD1). KLF1 has a dual role in the silencing of γ-globin genes: it activates BCL11A, a repressor of γ-globin gene expression, and it also activates the β-globin gene directly. BCL11A interacts with the GATA1, FOG1, and SOX6 erythroid transcription factors and with the NuRD deacetylase and remodeling complex to promote suppression of γ-globin gene expression. The nuclear receptors TR2/TR4 associate with corepressors DNMT1 and LSD1 as part of the DRED complex, a known repressor of embryonic and fetal globin genes in adults. MYB contributes to HbF regulation via activation of KLF1 (which activates BCL11A), activation of the DRED complex, and by modulating the number of F cells as part of its effect on erythroid differentiation kinetics and its pleiotropic effect on hematopoiesis.
Prospective targets for therapeutic induction of HbF
Identification of these genetic loci regulating HbF levels has revealed cellular pathways providing insights for new therapeutic targets for increasing HbF (Figure 1). The ideal target would be one that mimics and enhances the effect of the genetic variants that have been identified in modulating HbF levels, but does not affect other biological pathways. Targets identified in the emerging network of HbF regulation include MYB, KLF1, and BCL11A. Manipulating MYB to achieve an adequate therapeutic window could be problematic due to its pleiotropic role in hematopoiesis. KLF1 appears to be key in the γ-globin to β-globin switch; although KLF1 expression is almost exclusively restricted to erythroid cells, there would be difficulty in achieving specificity due to its broad array of erythroid activity.47 Reducing BCL11A activity is an attractive approach; proof-of-principle in HbF reactivation has already been demonstrated in a variety of systems, including cell lines, primary human erythroid cells, transgenics, and a mouse model of SCD with reversal of characteristic organ damage.56 However, BCL11A has essential roles in neuronal and B-lymphocyte development. Other promising targets include the DNA-methylating and histone-modifying enzymes. Proof-of-principle has already been provided by the effective in vivo HbF induction properties of 5-azacytidine and decitabine, both of which target the DNA methyltransferase DNMT1.2 Another attractive target is modulating expression of the orphan nuclear receptors TR2/TR4; one approach is inhibition of lysine-specific demethylase 1 (LSD1) via tranylcypromine (TCP), an antidepressive agent.57
From genetics to more precise risk prediction?
Although environmental factors are important in determining the clinical outcome of both SCD and β-thalassemia, it is evident that the genetic background of the affected individual imparts a substantial contribution to the clinical severity and response to iron chelators and hydroxyurea. Over the past 20 years, numerous association studies have been published and roles for many modifying factors and genes proposed, but the results are questionable because of the lack of replication. Furthermore, the genetic evidence generated about the phenotype has not been supported by functional assays or relevant models. Even if the variants are verified, they are likely to explain only a small fraction of the phenotype variation and, therefore, have limited predictive value and clinical utility.
One exception is HbF genetics. HbF levels in adults are highly heritable and controlled by 3 main loci, BCL11A, HBS1L-MYB intergenic polymorphisms, and Xmn1-HBG2. In healthy Europeans, the 3 loci explained more than 44% of the total variance in HbF levels (BCL11A 15.1%, HMIP 19.4%, and Xmn1-HBG2 10.2%),10 whereas in African patients with SCA, the 3 loci contribute from 16% (Tanzanians)24 to 20% (African Americans)23 of the HbF variance, with a corresponding reduction in frequency of acute painful crises. The impact of the HbF QTLs on HbF response and reduction in blood transfusion requirements has also been demonstrated in patients with β-thalassemia and HbE/β-thalassemia.12,38
The relatively large fraction (20%-50%) of phenotypic variation in HbF explained through a limited number of genetic loci is unique for a disease-relevant trait in humans compared with other traits and complex disorders, For example, in Crohn disease, 20% to 23% of disease risk can be explained by 71 QTLs.13 HbF could be the first human quantitative trait to be effectively predicted through genetic assay systems and its value tested to inform clinical or pharmacogenetic management (eg, response to HbF-activating agents).
Based on the current state of knowledge, the 3 HbF QTLs should be able to predict an individual's ability to produce HbF within a clinically meaningful confidence limit. Studies in Sardinia and France showed that a combination of the Xmn1-HBG2 site, SNPs in BCL11A and HMIP, together with α-globin genotypes can predict 75% to 80% of disease severity in β-thalassemia.37,39 In a cohort of 316 β0 thalassemia patients, delayed or absent transfusion needs were correlated with status of the 3 HbF QTLs and the α-globin genotype.58 In a recent study of patients with SCA, a panel of 14 SNPs comprising 6 from BCL11A and the rest from chromosome 11p15 encompassing the HBB gene complex and olfactory receptor gene region explained 23.4% of HbF variability.59 The value of HbF predictive genetics lies in the newborn and early childhood stage, when it may be possible to combine this knowledge with other laboratory and clinical parameters (eg, dactylitis in infants, leukocyte count, hemoglobin level, and TCD velocity) to predict severe disease outcomes such as stroke, frequent pain crises, and acute chest syndrome in SCA,25 thus facilitating early implementation of preventive therapy. In β-thalassemia, HbF predictive diagnostics could be integrated with information on β-globin and α-globin genotypes to better inform genetic counseling.
Conclusion
The generation of a personalized genetic risk score to inform prognosis and guide therapeutics has been a major driver underlying genetic association studies in numerous diseases and traits, including the β-hemoglobinopathies.60 A challenge that faces all GWAS is the translation of genetic risk variants into clinical utility. In this regard, predictive HbF genetics has 2 advantages: (1) the clinical impact of HbF is well documented and (2) a limited number of QTLs account for a substantial proportion of the trait variance. Already, identification of the loci regulating HBG production has provided new insights and leads for therapeutic HbF reactivation. A DNA assay-based diagnostic for the testing of one's ability to produce HbF could provide predictive information for those at risk of early complications. The success of predictive testing will depend on continued accurate identification of genetic and environmental factors and its ultimate clinical utility.
Acknowledgments
The author thanks Claire Steward for help in preparation of the manuscript and apologizes to the many investigators whose work could not be cited due to space limitations.
Disclosures
Conflict of interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Swee Lay Thein, King's College London School of Medicine, Molecular Haematology, The James Black Centre, 125 Coldharbour Lane, London SE5 9NU, United Kingdom; Phone: 44-20-7848-5443/5447; Fax: 44-20-7848-5444; e-mail: sl.thein@kcl.ac.uk.
