
Abstract
Over the past decade, the number of new therapies developed for the treatment of rare diseases continues to increase. The most rapid growth has been in the development of new drugs for oncology indications. One focus in drug discovery for oncology indications is the development of targeted therapies for select patient subgroups characterized by genetic alterations. The identification of these patient subgroups has increased in the past decade and has resulted in a corresponding increase in the development of new drugs for genetically defined patient subgroups. As an example of the development of new therapeutics for rare indications, I describe here the drug discovery efforts leading to the development of DOT1L inhibitors for the treatment of MLL-rearranged leukemia.
Introduction
A rare disease is defined as one affecting less than 200 000 patients in the United States. There are approximately 7000 rare diseases affecting more than 7% of the population.1 A drug developed for a rare disease may be indicated as an orphan drug. More than 2200 molecules are designated as orphan drugs, with between 30% and 40% being developed for oncology indications.1,2 In the United States, the increase in the number of drugs developed for rare diseases was stimulated after the introduction of the Orphan Drug Act (ODA) in 1983. The objective of the ODA was to stimulate research in rare diseases and the development of therapeutics to treat these rare diseases. The ODA created several incentives for the development of drugs including, but not limited to, a 7-year market exclusivity, research grants, and fast-track development and approval. Before 1983 and the ODA, only 10 products were approved in the United States for the treatment of rare diseases.1 After the establishment of the ODA, more than 400 drugs with orphan drug designation have obtained approval by the Food and Drug Administration (FDA), indicating that the ODA has indeed stimulated drug development activity for rare diseases.
Oncology drug discovery
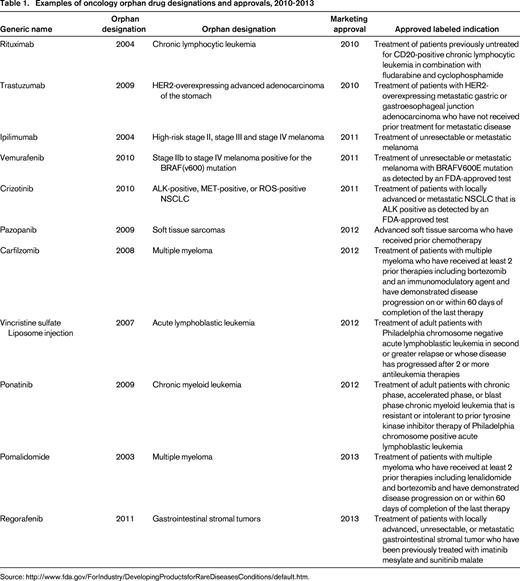
The therapeutic area with the greatest number of drugs with orphan designation and approval is oncology (Figure 1). This high number of orphan drugs in development for cancer is a result of a several factors, including the ability to identify and target the underlying oncogenic driver events in subpopulations of cancer patients. The large-scale “omics” efforts that are currently being undertaken to molecularly characterize hundreds of cancers from many cancer indications, such as The Cancer Genome Atlas (TCGA) project, have enabled the recent progress in identifying potential new oncogenic drivers.3 An example of this type of molecularly defined development is the approval of crizotinib in 2011 by the FDA for the treatment of patients with locally advanced or metastatic non-small-cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK)–positive as detected by an FDA-approved test. An ALK translocation resulting in activation of the ALK tyrosine kinase is detected in approximately 5% of cases of NSCLC. One of the potential opportunities of developing drugs in a defined patient population is that the clinical trials may require fewer patients, leading to more rapid approval. Indeed, the EML4-ALK translocation was identified in 2007 in NSCLC,4 and the “c-MET” inhibitor crizotinib was quickly repositioned and approved by the FDA in 2011 (Table 1). In addition, the identification of a defined patient population allows for early study of the new agent in the phase 1 setting. For example, the design for a phase 1 trial can include an expanded cohort of patients once the recommended phase 2 dose has been identified. The expanded patient cohort can include only the patients with the defined molecular target or alteration.

Orphan drug approval classified by therapeutic indication. The pie graph indicates the orphan drug approvals granted by the FDA between the years of 1983 and 2009. “Other” includes respiratory, intoxication, immunologic, psychiatric, musculoskeletal, gastrointestinal, dermatologic, ophthalmologic, hepatic/biliary, cardiovascular, and genitourinary indications.
Orphan drug approval classified by therapeutic indication. The pie graph indicates the orphan drug approvals granted by the FDA between the years of 1983 and 2009. “Other” includes respiratory, intoxication, immunologic, psychiatric, musculoskeletal, gastrointestinal, dermatologic, ophthalmologic, hepatic/biliary, cardiovascular, and genitourinary indications.
The number of drugs developed for patients with defined cancer subpopulations is likely to continue to increase due to the ongoing identification of genetic alterations in defined cancer subpopulations that have the potential to be oncogenic drivers of the cancer phenotype. This article focuses on the target validation and drug discovery efforts in a genetically defined subpopulation of leukemia that serves as an example of the discovery of new therapeutics in rare diseases. The drug discovery process can be broadly divided into 4 steps: (1) identification of the molecular mechanism driving the cancer subtype enabling the identification of a potential new drug target, (2) initial validation of the drug target in relevant preclinical model systems, (3) discovery and characterization of inhibitors for the drug target incorporating disease relevant cell-based and animal models leading to the selection of a drug candidate, and (4) investigational new drug (IND) enabling studies and entry into clinical testing in defined patient populations enabling rapid achievement of proof-of-concept in patients (Figure 2).

Drug discovery in rare diseases: steps from the bed to the bench and back again.
Drug discovery in rare diseases: steps from the bed to the bench and back again.
MLL-rearranged leukemia
The leukemia subgroup with translocations in the Mixed Lineage Leukemia (MLL) gene at 11q23 constitutes approximately 5% to 10% of acute myeloid leukemia and acute lymphocytic leukemia.5,6 More than 70% of infant acute lymphocytic leukemia patients have this alteration, and secondary leukemias that arise after treatment with topoisomerase inhibitors have translocation of the MLL gene. The 11q23 translocation in leukemia is a predictor of a poor outcome in patients.7 MLL is a histone methyltransferase and catalyzes methylation of histone H3 lysine 4 (H3K4). More than 50 translocation partners have been described for MLL and can be divided into 2 main classes.6,7 The first class consists of nuclear proteins that are involved in transcriptional activation/elongation. The second class consists of cytoplasmic proteins containing a dimerization domain that facilitates MLL homodimerization. The most common MLL translocation partners are in the first class and include AF4, AF9, ENL, and ELL. These nuclear proteins all share in common that they are components of transcriptional elongation complexes8-10 and retain the ability to recruit another histone methyltransferase, DOT1L, to the complex.11 DOT1L is a histone H3 lysine 79 (H3K79) methyltransferase and catalyzes mono-, di-, and trimethylation at this site.12 It is the only methyltransferase that has been shown to catalyze methylation of histone H3K79. Using the yeast 2-hybrid approach to identify interaction partners, DOT1L was shown to form a complex with the translocation partner AF10.13 The identification of the interaction of DOT1L with AF10 led to the hypothesis that DOT1L plays an important role in the development of MLL-rearranged leukemia.11 The MLL fusion protein loses the catalytic domain of MLL but retains the DNA-binding domain. Therefore, the MLL fusion protein binds to MLL target genes and recruits the DOT1L protein to these sites, resulting in increased H3K79 methylation at the promoter region of MLL target genes and enhanced expression of this set of target genes.14,15 Indeed, MLL rearranged leukemias have a characteristic gene expression profile compared with other leukemia subgroups.15,16 HOXA9 and MEIS1 are 2 examples of genes that are highly expressed in MLL-rearranged leukemias.5,17,18 Chromatin immunoprecipitation studies confirm the occupancy of the MLL fusion protein and enhanced H3K79 methylation at the promoter region of the gene set characteristic of MLL-rearranged leukemia.15,18 These studies support a critical role of DOT1L in development of MLL-rearranged leukemia.
Validation of DOT1L as a therapeutic target
To further support the role of DOT1L as a potential target for therapeutic intervention, several groups of investigators have carried out cell-based and in vivo validation studies. The initial studies performed confirmed that expression of the MLL fusion protein was sufficient to cause leukemia in mice.11 These studies also established the requirement of DOT1L enzymatic activity for leukemic transformation.11 Most studies have focused on the role of MLL-AF4 and MLL-AF9 fusion partners and dependency on DOT1L. These studies demonstrate that the MLL fusion is sufficient to cause transformation in mice and are dependent on DOT1L for the development of leukemia.13,18 These gain-of-function studies are supportive of the critical role of DOT1L in the transformative process, but do not address whether DOT1L is required for the maintenance of the transformed phenotype. Further support for DOT1L was demonstrated in genetic knock-down studies. These studies show that knock-down of DOT1L in cell lines containing the MLL translocation require DOT1L for viability.18 Further studies show that H3K79 methylation is critical for MLL-AF9–driven leukemogenesis using conditional knockout mice.13,18 The above studies support the selection of DOT1L as an important therapeutic target in MLL-rearranged leukemia.
Discovery of DOT1L inhibitors
The development of small-molecular inhibitors of DOT1L methyltransferase activity was initiated based on the supportive validation studies indicating a dependency on the enzymatic activity of DOT1L in the development of leukemia in the subset of patients whose leukemia possesses the MLL translocations.19 The dru-screening paradigm was based on the findings in in the initial validation studies and included the initial biochemical assay,20 followed by a cell-based biochemical screen evaluating H3K79 methylation in leukemic cell lines bearing the MLL translocation.19 Downstream pathway effects were evaluated by monitoring the effect on MLL fusion target gene expression, including HoxA9 and Meis1,21 and the effects on cell proliferation, apoptosis, and differentiation in leukemia cell lines with MLL translocations.19 The initial screen for inhibitors was a biochemical screen to identify small-molecule inhibitors of the methyltransferase activity of DOT1L. DOT1L is the only methyltransferase that catalyzes the methylation of histone H3K79. Cell-based assays to determine inhibitor effects on cellular H379 methylation levels were developed and used to determine cell biochemical activity and inhibitor selectivity. Additional assays were developed to evaluate MLL fusion target gene expression and cell viability. The cell-based studies were performed in human cell lines harboring the MLL translocation. The established MLL-rearranged cell lines expressed the gene set characteristic of primary MLL-rearranged leukemia. It is critical to initiate these studies in preclinical model systems that most accurately represent the disease under study. Using model cell lines that accurately reflect the patient population intended to be treated in the initial clinical trial streamlines the drug discovery program and improves the translation of the preclinical studies in model systems to the clinical trials. Additional assays were developed to determine inhibitor effect on in vitro and in vivo MLL-rearranged tumor models as appropriate. At an early point in the drug discovery program, it is also important to develop the biomarker strategy for the program. This strategy includes both pharmacodynamic and patient selection biomarkers. There is a clear strategy for DOT1L for the patient selection biomarker based on the presence of MLL translocation. In addition, pharmacodynamic biomarkers were identified in the validation studies and include inhibition of histone H3K79 methylation and MLL fusion target gene expression modulation. The pharmacodynamic biomarkers will aid in the selection of the dosage regimen that will be used in the clinical studies. The development of the pharmacodynamics and patient selection biomarker assays during the drug development phase enables incorporation of these assays in the first in human (FIH) phase 1 clinical trials.
Highly potent and selective DOT1L inhibitors were developed using the assays described above. The initial inhibitor EPZ4777 demonstrated sub- to low-nanomolar inhibition of DOT1L enzymatic activity, inhibition of MLL fusion target gene expression, and induction of apoptosis in MLL-rearranged leukemia cells.18,19 The DOT1L inhibitors selectivity inhibited DOT1L enzymatic activity in biochemical assays and in cell-based assays, as evidenced by selective inhibition of cellular histone H3K79 methylation. Further inhibitors, including EPZ5676, were developed and evaluated in biochemical, cell-based assays and in vivo studies.22 To evaluate whether DOT1L inhibitors selectively inhibited MLL-rearranged leukemias, the inhibitors were evaluated in a panel of cell lines that did and did not have the MLL rearrangement.19 These studies indicate that the DOT1L inhibitor is active in MLL-rearranged leukemic cells and supported further in vivo evaluation of these inhibitors in relevant models of MLL-rearranged leukemia. The DOT1L inhibitor EPZ-5676 demonstrated significant antitumor activity.19 These studies support the further development of the DOT1L inhibitor for the treatment of MLL-rearranged leukemia. Initial phase 1 clinical trials have been initiated with the primary purpose of determining what dose of EPZ-5676 can be given safely to patients with hematologic malignancies (www.clinicaltrials.gov identifier NCT01684150). The study has 2 phases. In the first phase, the maximally tolerated dose of EPZ-5676 will be established. Once that is established, a second phase of the study will further evaluate the safety of EPZ-5676 and assess its antileukemia activity in patients with MLL-rearranged leukemia.
Current challenges and future perspectives
In summary, the number of opportunities to develop new therapies for rare diseases including cancer is likely to increase as we continue to understand the drivers of disease in defined patient subpopulations. One of the major challenges for the discovery of new drugs in defined cancer subtypes is incorporating the relevant preclinical models that accurately reflect the disease under study. Model systems are often not available and the generation of appropriate models can require long lead times, thus lengthening the drug discovery timeline. The lack of predictive preclinical models dramatically lowers the probability of successfully transitioning the therapeutics into patients. Another major hurdle is the development of biomarker assays for patient selection. As we move to defined patient populations in early clinical development, it is essential to incorporate quantitative biomarker assays into our preclinical and clinical studies. A third issue is the ability to identify and recruit patients to studies in a timely fashion. These issues may be offset with our ability to conduct smaller studies and obtain potentially higher response rates when using selected patients.23 Recent successes in treating defined patients subsets [eg, ALK in NSCLC and Raf (V600E) in melanoma] have demonstrated that the approach of targeting specific alterations in defined cancer indications can lead to patient benefit and approval of new drug entities.
Disclosures
Conflict-of-interest disclosure: The author has equity ownership in Epizyme and is employed by Sanofi. Off-label drug use: None disclosed.
Correspondence
Victoria M. Richon, PhD, Sanofi Oncology, 270 Albany Street, Cambridge, MA 02139; Phone: 617-665-4301; e-mail: victoria.richon@sanofi.com.
