
Abstract
Waldenström macroglobulinemia (WM) is a rare lymphoproliferative disorder characterized by the presence of lymphoplasmacytic cells in the BM and IgM monoclonal protein in the serum. The origin of the malignant clone is thought to be a B cell arrested after somatic hypermutation in the germinal center and before terminal differentiation to plasma cells. In this review, recent advances in the genetic and epigenetic regulators of tumor progression are discussed. Risk factors include IgM-monoclonal gammopathy of undermined significance, familial disease, and immunological factors. The clinical manifestations of the disease include those related to clonal infiltration of the BM, lymph nodes, and, rarely, other sites such as pulmonary or CNS infiltration (Bing-Neel syndrome). Other manifestations are related to the IgM monoclonal protein, including hyperviscosity, cryoglobulinemia, protein-protein interactions, Ab-mediated disorders such as neuropathy, hemolytic anemia, and Schnitzler syndrome. IgM deposition in organs can lead to amyloidogenic manifestations in WM. The diagnostic workup for a patient with WM and rare presentations of WM are described herein. Prognosis of WM depends on 5 major factors in the International Staging System, including age, anemia, thrombocytopenia, β-2 microglobulin, and IgM level. The differential diagnosis of WM includes IgM-multiple myeloma, marginal zone lymphoma, mantle cell lymphoma, and follicular lymphoma.
Introduction
Waldenström macroglobulinemia (WM) is a distinct B-cell lymphoproliferative disorder primarily characterized by lymphoplasmacytic cells infiltrating the BM, along with demonstration of an IgM monoclonal gammopathy in the serum.1–4 In 1944, Jan Gosta Waldenström described the entity by describing the case of 2 patients with lymphadenopathy, oronasal bleeding, anemia, thrombocytopenia, elevated erythrocyte sedimentation rate, high serum viscosity, and infiltration of the BM by lymphoid cells. Dr Waldenström interpreted the symptoms to be related to hyperviscosity attributed to the presence of an abnormally elevated high-molecular-weight serum protein, as he showed by paper electrophoresis, which subsequently was demonstrated to be a monoclonal pentameric IgM.
According to the Revised European American Lymphoma (REAL) and World Health Organization (WHO) classifications, WM is classified as a lymphoplasmacytic lymphoma (LPL).3,4 In the United States each year, there are 1500 new cases diagnosed. The overall incidence of WM is approximately 3 per million persons per year.5,6 Based on 11 population-based registries within the United States, the age-adjusted rates for men and women are 3.4 and 1.7 per million, respectively. When segregated by age, the rates increased sharply: 0.1 per million ages < 45 years and 36.3 per million ages 75+ years. In addition, the rates for whites are clearly higher than those for blacks.5 Accurate estimates of the prevalence of WM are complicated by the ill-defined overlap between IgM-monoclonal gammopathy of undermined significance (IgM-MGUS) and asymptomatic WM.
In this review, the diagnostic criteria of WM and the clinical manifestations of the disease, including diagnostic workup and rare presentations of WM, are defined and a discussion of other conditions that are in the differential diagnosis of WM are discussed. Therapy options are not covered in this review, but have been extensively discussed previously.7,8
Biology of WM
To diagnose a disease, it is critical to start with the origin of the clonal cells and their biological characteristics. The origin of the malignant clone is thought to be a B cell arrested after somatic hypermutation in the germinal center and before terminal differentiation to plasma cells.9 This indication has been confirmed by results of the analysis of the nature and distribution of somatic mutation in Ig heavy- and light-chain variable regions obtained from patients with WM, indicating that WM may originate from an IgM+ and/or IgM+IgD+ memory B cell, with a deficiency in the initiation of the switching process.
Cytogenetic findings and copy number variations.
Several studies have been conducted to investigate and define the possible role of genetic abnormalities in the pathogenesis of WM. The most common cytogenetic abnormalities identified by FISH analysis is the deletion of the long arm of chromosome 6, which was reported in up to 55% of patients with WM in one study.10 Other studies demonstrated the presence of other cytogenetic abnormalities, including trisomy 4, trisomy 5, monosomy 8, and deletion of the long arm of chromosome 20.10 Among the genes that are deregulated in WM, BLIMP-1 is one of the candidate tumor suppressor genes in 6q21 that is under study because it regulates the transition of mature B cells to terminally differentiated plasma cells.11
Array-based comparative genomic hybridization approaches showed that 83% of WM patients have chromosomal abnormalities.12 Gain of 6p was the second most common abnormality (17%), and its presence was always concomitant with 6q loss. A minimal deleted region, including MIRN15A and MIRN16-1, was found on 13q14 in 10% of patients. Biallelic deletions and/or inactivating mutations were observed with uniparental disomy in TNF receptor-associated factor 3 (TRAF3) and TNFα-induced protein 3 (TNFAIP3), 2 negative regulators of the NF-κB signaling pathway.
Genomic sequencing
Whole-genome sequencing of lymphoplasmacytic cells from 30 WM patients was performed with paired normal tissue sequencing for 10 patients and presented in abstract form at the American Society of Hematology meeting in 2011.13 A recurring sequence variant in chromosome 3p22.2 was identified, which resulted in a single nucleotide change in the myeloid differentiation primary response (MYD88) gene with a predicted nonsynonymous change at amino acid position 265 from leucine to proline (L265P). This MYD88 L265P mutation occurred in 90% of the WM samples and was not observed in samples of multiple myeloma (MM), IgM-MGUS, or healthy subjects.
Epigenetics
Multilevel genetic characterization of WM is required to improve our understanding of the underlying molecular changes that lead to the initiation and progression of this disease.14 miRNA-expression profiling revealed a specific signature characterized by increased expression of miRNA-363*/-206/-494/-155/-184/-542-3p, and decreased expression of miRNA-9*. miRNA-155 was found to be a critical regulator of tumor proliferation and growth of WM cells in vitro and in vivo.14 In addition, increased expression of miRNA-206 and reduced expression of miRNA-9* regulated histone deacetylases and histone acetyl transferases, indicating that these miRNAs may play a role in regulating histone acetylation in WM.15
Risk factors for developing WM
The causes of WM or LPL are poorly understood. Risk factors include the presence of pre-existing IgM-MGUS, familial history of WM or other B-cell malignancies, and immunological factors.
IgM-MGUS.
The main risk factor for the development of WM is pre-existing IgM-MGUS, which confers a 46-fold higher relative risk of developing WM than for the general population.16 The prevalence of all Ig isotypes of MGUS is approximately 3% in a white population over the age of 50 years based on the Olmsted County data collected by Kyle et al.16 The estimated incidence of IgM-MGUS is 0.55% in white subjects over the age of 50.16 The study by Kyle et al in 213 patients diagnosed with IgM-MGUS showed that the relative risk of progression to WM, lymphoma, and amyloidosis was 262, 14.8, and 16.3, respectively, indicating that IgM-MGUS progresses most commonly to WM; the cumulative incidence of progression was 10% at 5 years, 18% at 10 years, and 24% at 15 years, respectively.16 The serum monoclonal protein and serum albumin concentrations at diagnosis were the only risk factors for progression. Our recent study showed that elevated serum free light chain (with a cut off of 60 mg/L) is reflective of tumor burden and can be used to differentiate IgM-MGUS from symptomatic WM.17 Similarly, 6q deletion is not seen in IgM-MGUS, but it is observed in 30%-50% of patients with WM.18
Familial WM.
WM represents a sporadic disease in most cases; however, various reports indicate a high familial incidence.19,20 In one study of 257 WM patients, 19% had at least one first-degree relative affected with WM or another B-cell disorder, including non-Hodgkin lymphoma, MM, chronic lymphocytic leukemia MGUS, acute lymphoblastic leukemia, and Hodgkin lymphoma.19,20 In a large study from Sweden of 2144 LPL/WM patients, 8279 population-based matched controls and linkable first-degree relatives of patients (n = 6177) and controls (n = 24 609), there was a significantly increased risk of first-degree relatives of LPL/WM patients in developing LPL/WM, other subtypes of non-Hodgkin lymphoma, and MGUS, but not Hodgkin lymphoma or MM, compared with first-degree relatives of controls.21–23 The excess risks were similar among parents, siblings, and offspring, which suggests a dominant or codominant gene effect rather than recessive genes. The strongest evidence of linkage in a genome-wide linkage analysis of 11 high-risk WM families was found on chromosomes 1q and 4q. Other locations suggestive of linkage were found on chromosomes 3 and 6.
Immunological risk factors.
Emerging studies support the role of immune-related factors in the pathogenesis of LPL/WM.21,23,24 In a study of 103 WM patients and 272 unaffected family members, familial WM patients were more likely than unaffected relatives to report a history of autoimmune disease and infections. Familial WM patients were also more likely to report exposure to farming, pesticides, wood dust, and organic solvents compared with unaffected family members.
Diagnosis
Diagnostic criteria for WM are defined by the presence of a serum IgM component accompanied by BM infiltration of small lymphocytes (with plasmacytoid or plasma cell differentiation).4 Attempts to better define the diagnosis of WM have been made by the WHO Lymphoma Classification, the consensus group formed at the Second International Workshop on Waldenström's Macroglobulinemia and the Mayo Clinic.1–4 However, the respective definitions of the diagnostic criteria for WM by these groups are not identical. All groups recognize WM as an LPL associated with an IgM monoclonal protein in the serum. The WHO definition includes lymphomas other than LPL and also allows the monoclonal protein to be IgG or IgA. In contrast, the Second International Workshop on Waldenström's Macroglobulinemia restricts the diagnosis exclusively to cases with LPL and an IgM monoclonal protein. That workshop also eliminated the requirement for either a minimum amount of BM involvement by LPL or a threshold concentration of IgM in the serum to fulfill the diagnosis and allows any detectable amount of either. In contrast, Mayo Clinic criteria require at least 10% BM involvement to differentiate IgM-MGUS from LPL. Furthermore, in regard to pathologic features, the WHO criteria focus predominantly on nodal involvement, whereas studies at Mayo Clinic indicate that in most cases of WM, the LPL is a BM-based disease.1–4
BM involvement in WM is characterized by a monotypic lymphocytic component with a characteristic immunophenotype of high levels of surface CD19, CD20, and Ig light chain expression.1–4 The intratrabecular pattern of BM infiltration, which may be diffuse, interstitial, or nodular, is commonly found in WM patients, whereas a solely paratrabecular pattern is rare and should raise suspicion of follicular lymphoma. Dutcher bodies are frequently present in the plasma cells. An increased number of mast cells close to lymphoid aggregates are commonly found in WM.
The BM infiltration should routinely be confirmed by immunophenotypic studies showing the following profile: sIgM+, CD19+, CD20+, CD22+, CD79+. The lymphoid component typically lacks CD10. In approximately 40% of patients, the lymphocytes show some degree of CD5 expression; however, these patients usually do not show the strong expression of this Ag associated with chronic lymphocytic leukemia/small lymphocytic lymphoma or mantle cell lymphoma. In comparison, the plasmacytic component expresses the same Ig light chain as the lymphocytic component, is positive for CD138 (particularly when assessed by immunohistochemistry), and shows diminished expression of B-cell–associated Ags such as CD19, CD20, and PAX5. Typically, the LPL cells are positive for surface IgM, but on the basis of the WHO criteria, they may express any Ig isotype. In patients with isotype switch, the phenotype of the plasma cells closely resembles that of myelomatous plasma cells, with strong CD38 and CD138 coexpression and complete lack of CD19. WM cells have also been shown to be CD25+, CD27+, CD75–, FMC7+, Bcl2+, and Bcl6−.1–4 Although not completely specific for LPL, del(6)(q21) is the most common genetic abnormality detected by FISH, being seen in 40%-50% of patients. This genetic abnormality is rarely seen in other lymphoproliferative or plasmaproliferative malignancies. With the recent findings of MYD88 L265P mutation in 90% of cases of WM, a clinical test may be developed soon to aid in the diagnosis of this disease.
Clinical features
Patients with WM can present with an extensive range of signs and symptoms.1–4 The majority present with signs and symptoms related to the monoclonal serum protein and/or to the tumor infiltration. Frequent clinical presentations are related to cytopenias, specifically anemia associated with the replacement of tumor cells in the BM. Patients may also present with symptoms related to hyperviscosity. Approximately 20% of patients will experience hepatosplenomegaly and lymphadenopathy, and some patients may present with B symptoms including night sweats, fever, and weight loss. Other common manifestations include neuropathy, cryoglobulinemia, skin rash (Schnitzler syndrome),25 cold-agglutinin hemolytic anemia, and amyloidosis.26,27
Spectrum of the disease: IgM-MGUS, asymptomatic WM, and symptomatic WM
WM presents with a progression from IgM-MGUS to smoldering WM and, finally, symptomatic WM.28,29 IgM-MGUS is defined as an IgM < 3 g/dL and < 10% lymphoplasmacytic cells in the BM. Smoldering WM is defined as an IgM > 3 g/dL or more than 10% LPL cells in the BM in the absence of any symptoms related to WM. The cumulative probability of progression of smoldering WM to symptomatic WM, amyloidosis, or lymphoma is 6% at 1 year, 39% at 3 years, and 59% at 5 years, respectively.29 However, the criteria used for IgM-MGUS and smoldering WM are not widely adopted in the WM literature but may be useful in differentiating time to progression in patients with asymptomatic disease. Another classification that is more frequently used is asymptomatic and symptomatic WM based on the consensus recommendations that patients with any level of IgM monoclonal protein or presence of lymphoplasmacytic cells in the BM should be diagnosed with WM4 (Table 1).
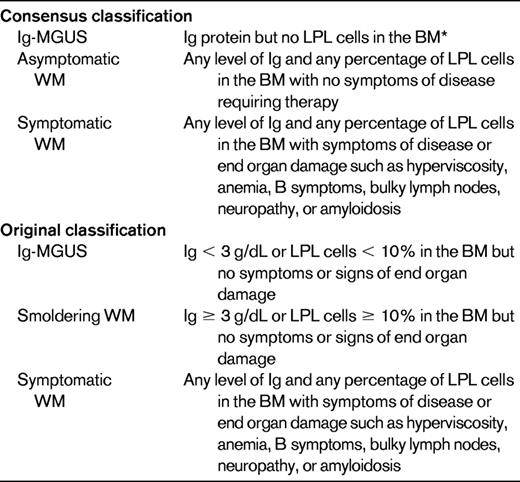
Patients with asymptomatic macroglobulinemia are those with any level of monoclonal protein level and any level of infiltration of the BM with lymphocytes and plasma cells, but with no constitutional symptoms, hepatosplenomegaly, lymphadenopathy, anemia, or thrombocytopenia. Such patients should be recognized and not treated because they may remain stable for many years. In an analysis of 31 asymptomatic patients, the median time to disease progression was 6.9 years.3 Prognostic factors for early progression were hemoglobin < 11 g/dL, β2-microglobulin > 3.0 mg/dL, and serum monoclonal protein > 3 g/dL. The median time to progression was expected to be 10 years with no adverse features (55% of patients), 2 years with 1 abnormality (29% of patients), and 0.5 years with 2 or more abnormalities (16% of patients).
Diagnostic workup and clinical manifestations of symptomatic WM
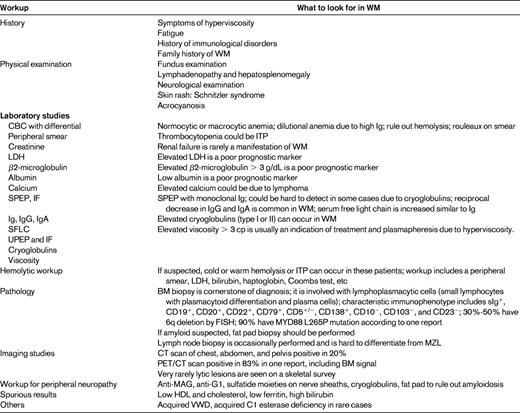
The factors that are critical in the history and physical examination of WM patients are highlighted in Table 1. The clinical presentations of WM can be divided into those related to clonal cell infiltration and those related to the IgM component:
Tissue infiltration by clonal cells.
Clonal cells predominantly infiltrate the BM, lymph nodes, liver, and spleen. However, WM cells can also infiltrate other organs such as the lungs or CNS in rare cases.
The BM infiltration results in cytopenias and progressive anemia. A complete blood count may show normocytic or macrocytic anemia. The most common causes of anemia are BM involvement and anemia of chronic disease. Hemolytic anemia and immune thrombocytopenia (ITP) can occur in some patients with WM. Hemolytic anemia can be warm- or cold-agglutinin hemolytic anemia as described in “Ab activities.” Exclusion of other causes of anemia is critical, especially if this is the only reason to treat a patient with WM. Dilutional anemia is very common with elevated levels of IgM. Many patients have low levels of iron, so iron deficiency anemia should be investigated and, if present, treated adequately. As described in “Spurious laboratory measurements due to precipitation or interference by the IgM paraprotein,” low levels of ferritin may be due to interference of the IgM monoclonal protein with adequate detection of the serum level of ferritin.30
Lymphadenopathy and organomegaly are reported to occur in approximately 15%-20% of patients with WM, and computed tomography (CT) scans have been recommended for followup of disease response in patients with positive findings before therapy.27,31,32 A range of imaging findings have been previously described in the literature in patients with WM, including BM signal abnormalities on magnetic resonance imaging (MRI), skeletal uptake on bone scintigraphy, and lymphadenopathy on CT.33,34 The prognostic significance of the presence of imaging findings in WM is unclear, although one large study showed an adverse prognosis in patients with organomegaly.35
The use of combined fluorescein di-β-D-galactopyranoside (FDG)-positron emission tomography (PET)/CT imaging showed positive findings in 83% of patients with WM, unlike prior studies using conventional imaging, which found that only 20% of WM patients have lymphadenopathy or hepatosplenomegaly. Moreover, 43% of patients had abnormal BM uptake on FDG-PET imaging that can potentially help in the assessment of their tumor load, especially with heterogenous sampling of the BM. The presence of lytic lesions is very rare in WM, but can still occur in some patients.
Tissue infiltration of other organs by neoplastic cells is rare and can involve various organs and tissues. Pulmonary involvement in the form of masses, nodules, diffuse infiltrate, or pleural effusions is relatively rare, but has been described with WM. Cough is the most common presenting symptom, followed by dyspnea and chest pain.3 Malabsorption, diarrhea, bleeding, or obstruction may indicate involvement of the gastrointestinal tract at the level of the stomach, duodenum, or small intestine. Direct infiltration of the CNS constitutes the rarely observed Bing-Neel syndrome, which is characterized clinically by headache, vertigo, ataxia, diplopia, impaired hearing, and eventually coma. Ocular, periorbital, and retroorbital infiltration may be observed rarely.3,8,27
Clinical manifestations related to the monoclonal IgM component.
As shown in Table 2, the monoclonal IgM can cause several clinical manifestations through different mechanisms of action attributable to its physiochemical properties, nonspecific interaction with other proteins, autoimmune activity, and ability to deposit in tissue.
Physicochemical effects of the IgM
Serum protein electrophoresis and immunofixation are critical in establishing the diagnosis and characterizing the IgM monoclonal protein. Densitometry should be used to determine IgM levels, the measurement of which may be affected by the presence of cold agglutinin or cryoglobulins. For this reason, testing for cold agglutinin and cryoglobulins should be performed at diagnosis and, if present, serum samples should be analyzed under warm conditions. Serum Ig levels should be followed in the same institution because they can vary considerably between centers. There is no correlation between the level of IgM in the peripheral blood and the percentage of BM involvement. Although Bence-Jones proteinuria is frequently present in WM patients, it rarely exceeds 1 g/24 hours (only 3% of cases). Therefore, urine protein electrophoresis is not routinely performed in many patients with WM. The serum free light chain assay has been widely used in other plasma cell dyscrasias. It has also been shown in a small study to be a useful marker in following disease progression and response in WM.36
Hyperviscosity syndrome.
Due to the presence of elevated serum IgM levels, WM patients may present with clinical features from hyperviscosity. Despite representing the most common and typical feature of WM, hyperviscosity is observed in < 15% of patients at diagnosis.3,8,27,37 Symptoms occur when the serum viscosity reaches 4-5 centipoiesis (cp), which usually correlates with a serum IgM level ≥ 3000 mg/dL. Common symptoms include headache, blurred vision, and oronasal bleeding. A funduscopic examination is recommended in these patients because it appears to be an excellent indicator of hyperviscosity, showing dot-, blot-, and flame-shaped hemorrhages in the macular area accompanied by markedly dilated and tortuous veins with focal constrictions, resulting in the so-called “venous sausaging.” The increased blood viscosity and expanded plasma volume arising from increased osmotic pressure can aggravate congestive heart failure.
Type I cryoglobulinemia.
Cryoglobulins (type I) may be observed in 7%-20% of WM patients, but are symptomatic in 5% or less. Symptoms and signs result from the impaired blood flow in small vessels and include Raynaud phenomenon, acrocyanosis, arthralgias, purpura, and skin ulcers.3,8,27,37 If suspected, measurement of cryoglobulins using a warm bath collection should be ordered. As described in “Physicochemical effects of the IgM,” cryoglobulinemia can lead to underestimation of the serum IgM levels.
Ab activities
Monoclonal IgM may exert its pathogenic effects through specific recognition of autologous Ags such as nerve constituents, Ig determinants, and RBC Ags.
Neuropathy.
Peripheral neuropathy has been reported in 24% of WM patients.3,8,27,37 Anti–myelin-associated glycoprotein (anti-MAG) Ab has been described as one of the factors responsible for the demyelinating neuropathy found in WM. Moreover, IgM deposits at the site of MAG localization may be demonstrated after nerve biopsy. Other Abs could be anti-ganglioside M1 and anti-sulfatide IgM Abs. The absence of autoantibodies does not exclude the diagnosis of IgM-related neuropathy, because other myelin-associated Ags may be present that are currently not evaluable. More rarely, nerve damage may be mediated by amyloid deposits or by neoplastic cell infiltration of nerve structures. In addition, cryoglobulinemia can present with neuropathy in these patients.
The most commonly encountered symptomatic neuropathy in WM is distal and symmetrical, affecting both motor and sensory functions; it is slowly progressive with a long period of stability. Most patients present with sensory manifestations (paresthesias, aching discomfort, dysesthesias, or lancinating pains), imbalance and gait ataxia because of lack of proprioception, and leg muscle atrophy in advanced stages.
Type II cryoglobulinemia.
The monoclonal IgM may have an autoantibody activity against the FC portion of IgG, acting as a monoclonal rheumatoid factor.3,8,27,37 The cryoprecipitating property results from the size and limited solubility of the IgM-IgG immune complex. The systemic vasculitis that characterizes the disease appears to be caused by deposition of immune complexes on the walls of small vessels and subsequent activation of the complement cascade. The clinical features are purpura, arthralgias, weakness, liver involvement, renal involvement (cryoglobulinemic glomerulonephritis), peripheral neuropathy, and widespread vasculitis.
Cold agglutinin hemolytic anemia.
Monoclonal IgM may present with cold agglutinin activity and it can recognize specific RBC Ags at temperatures below physiological, producing chronic immune hemolytic anemia.3,8,27,37 This disorder occurs in < 10% of WM patients and is associated with cold agglutinin titers > 1:1000 in most cases. The monoclonal component is usually an IgMκ and reacts most commonly with I/i Ags, with complement fixation and activation. Mild chronic hemolytic anemia can be exacerbated after cold exposure, but rarely does hemoglobin decrease below 70 g/L. The agglutination of RBCs in the cooler peripheral circulation also causes Raynaud syndrome, acrocyanosis, and livedo reticularis.
ITP.
ITP can occur in patients with WM.1–4 ITP should be differentiated from other causes of thrombocytopenia due to BM infiltration or hypersplenism. ITP patients may benefit from rituximab or treatment of the underlying WM, and in some cases splenectomy may be indicated.
Schnitzler syndrome.
Schnitzler syndrome is a rare disorder characterized by a chronic urticarial rash and monoclonal gammopathy accompanied by intermittent fever, arthralgia or arthritis, bone pain, and lymphadenopathy.38 The etiology of Schnitzler syndrome remains unknown. Several hypotheses have been proposed, most of which suggest the involvement of autoreactive Abs.
Other Ab-mediated manifestations.
Acquired VWD leading to easy bleeding and bruising can occur in some patients with WM.39 VWD is usually associated with autoimmune or clonal proliferation disorders and, although the precise mechanism of acquired deficiency of VWF is poorly understood, the most likely candidate mechanisms are that Abs inactivate or form a complex with immunologic or functional sites on VWF, or that VWF multimers are absorbed selectively by malignant cells. Similarly, acquired C1 esterase deficiency can be observed in rare cases, leading to angioedema.40
Tissue deposition
Manifestations related to amyloidogenic properties of IgM
The deposition of the monoclonal light chain as amyloid deposits can also occur in patients with WM.26 In a series of amyloid patients, approximately 2% had an IgM monoclonal protein and, of these, 21% had associated WM. The most commonly affected organs are the heart (44%), peripheral nerves (38%), kidneys (32%), soft tissues (18%), liver (14%), and lungs (10%). Patients with amyloid and WM could be diagnosed by biopsy of the subcutaneous fat or BM. Rare cases of secondary amyloidosis have also been reported in WM, in which fibrils are derived from the acute-phase reactant serum amyloid A protein with subsequent deposition in tissues. These patients primarily present with nephrotic syndrome and gastrointestinal involvement.
Protein-protein interactions
Monoclonal IgM component may interact with circulating proteins or with proteins expressed on cell membranes, including fibrinogen and factors V, VII, and VIII, which are responsible for abnormalities in bleeding and clotting times.
Spurious laboratory measurements due to precipitation or interference by the IgM paraprotein
Paraproteins can interfere in measurements when they form precipitates during the testing procedure. This can lead to artifactually increased total bilirubin concentration and an artifactually low high-density lipoprotein in a patient with a monoclonal IgM paraprotein.41 Similar findings can be observed with low ferritin and transferrin. Low levels of ferritin may be due to interference of the IgM monoclonal protein with adequate detection of the serum level of ferritin.30
Survival and prognosis of WM
The median overall survival of patients with WM is 5-6 years, and the median disease-specific survival of symptomatic WM is 11.2 years.35 Although WM represents an indolent disease, a wide variability in prognosis can be observed; therefore, it is important to define prognostic factors. Several studies have shown that poor prognosis of patients with WM is associated with the following factors: advanced age, high β2-microglobulin, cytopenias, low albumin, serum IgM monoclonal protein, and organomegaly.35,42–45 The most widely used prognostic system is the International Prognostic Scoring System (ISS-WM).46 The scoring system is based on 5 risk factors and the survival of the patients at 5 years. Using the ISS-WM, patients with low-risk disease were shown to have a 5-year survival of 87%, whereas patients with high-risk disease had a 5-year survival of only 36%.
Differential diagnosis
Diseases that have similar characteristics to WM include IgM-MM, marginal zone lymphoma (MZL), mantle cell lymphoma, and other IgM-secreting lymphomas. Table 3 lists the characteristics differentiating these diseases from WM.

IgM-MM
IgM-MM is differentiated from WM by the presence of a purely plasmacytic infiltrate in the BM, even if these plasma cells express CD20+ markers by flow cytometry.1–4 In addition, IgM-MM is characterized by the presence of osteolytic lesions and renal insufficiency more frequently than in WM. Unlike patients with MM, renal dysfunction is rare in WM, but can occur in some patients with predominant light chain secretion, amyloidosis, and cryoglobulinemia. Cytogenetic characteristics of MM such as 13q deletion, 11:14, or 4:14 translocations may help to distinguish MM from WM. The IgH translocations are critical in the differentiation of WM from MM. A recent study defined IgM-MM as a clonal plasma cell–proliferative disorder characterized by an IgM monoclonal protein (regardless of size), 10% or more plasma cells on BM biopsy, plus the presence of lytic bone lesions and/or translocation t(11;14).47 In addition, recent findings of mutations in MYD88 that is present in WM but not in MM may also help to differentiate the 2 entities. The presence of lytic lesions should not be the only criterion to differentiate MM from WM, because rare cases of WM can present with lytic lesions as described above.
MZL
Like WM, MZL is composed of lymphocytes, lymphoplasmacytoid cells, and tumoral plasma cells with a surface IgM–positive/cytoplasmic IgM.1–4 In some cases, it is difficult to differentiate WM from MZL, specifically splenic MZL (SMZL). The nodal MZL is characterized by variable BM infiltration and a typical localized or generalized peripheral adenopathy with marginal zone and interfollicular areas with marginal zone, monocytoid, and small B cells; sometimes a plasma cell differentiation may be observed. Conversely, SMZL mostly involves the spleen, with small round lymphocytes that replace germinal centers and infiltration of red pulp. Splenomegaly is very common, with rare involvement of adenopathies or extranodal sites. For immunophenotyping, WM and SMZL consistently express pan-B-cell markers (ie, CD19, CD20, CD22, and surface Ig). However, there are differences in the k/l ratio (1.2:1 for SMZL and 4.5:1 for WM) and in some markers such as CD22 and CD11c, which are overexpressed in patients with SMZL compared with patients with WM, whereas CD25 are more frequently positive in WM (88% vs 44%). The CD103 Ag is always negative in WM, whereas it is positive in 40% of SMZL cases. The mAb FMC7 is usually positive in both entities: heterogeneous in WM but homogeneous in SMZL. The combination of CD25 and CD22 could differentiate between WM and SMZL. The principal molecular abnormality in WM is 6q deletion (30%-50% in WM), whereas in SMZL, the most common abnormalities are loss of 7q (19%) along with +3q (19%) and +5q (10%). Again, MYD88 mutation analysis may prove to be a useful marker to distinguish WM from other similar entities.
Mantle cell lymphoma.
Mantle cell lymphoma may be differentiated from WM because of the BM infiltration by monomorphous, small-medium lymphoid cells with irregular nuclei. In addition to the BM, it usually involves lymph nodes and extranodal sites such as the gastrointestinal tract and the spleen and in almost all cases of mantel cell lymphoma, there is t(11;14) (q13;q32).1–4
Follicular lymphoma.
Follicular lymphoma differs from WM because of the BM infiltration of small, cleaved cells that is usually paratrabecular. In addition, cytogenetic analysis shows rearrangement of Bcl-2 in 70%-90% of the cases.1–4
Treatment
This review will not address treatment options for WM, which are addressed in prior reviews.8,48 Asymptomatic patients with WM do not require therapy.49 Initiation of therapy is not based on the level of the IgM protein, but instead is based mainly on signs and symptoms. The most common reason for the initiation of therapy is anemia. Other causes include hyperviscosity symptoms, cytopenias, evidence of disease transformation, and significant neuropathy, adenopathy or hepatosplenomegaly, and amyloidosis.37,44,49
There are no US Food and Drug Administration (FDA)–approved therapeutic agents or standard of therapy for the treatment of WM.49 Many factors should be considered in making the decision for therapy, including the age of the patient, the presence of cytopenias, the rate of disease progression, the level of IgM protein, and the presence of neuropathy.
The recommendations for upfront therapy for WM include alkylating agents, nucleoside analogs, the mAb rituximab, and bortezomib-based combinations.48,49 For patients with relapsed disease, the use of alternate frontline agents, reuse of a frontline agent, use of combination myelotoxic chemotherapy, and use of thalidomide, bortezomib, or other novel therapeutic agents are recommended.48,49 High-dose chemotherapy with autologous stem cell rescue in primary refractory or relapsed disease should be considered for eligible patients. However, allogeneic and “nonmyeloablative allogeneic” transplantations should be approached cautiously because of the associated high mortality and/or morbidity risks, and should be undertaken only in the context of a clinical trial.48,49
Many novel therapeutic agents are being tested in clinical trials for WM. Given the paucity of large clinical trials in WM, especially phase 3 clinical trials, the establishment of a standard treatment regimen that can be used for the comparison of the response obtained with these trials has become challenging. For more extensive recommendations on therapy and updates of new agents currently used in WM, please refer to other reviews on this topic.7,8
Treatment of IgM-related neuropathy includes the use of immunoglobulins with corticosteroids, plasma exchange, and therapeutic interventions to decrease the IgM protein level.50–52 The most common therapy used is rituximab alone or in combination.53–55 This strategy has shown clinical benefit and reductions in the titers of Abs. Patients with severe IgM-related neuropathy may benefit from cytotoxic chemotherapy alone or in combination with rituximab, such as cyclophosphamide and rituximab.8 Other classes of novel agents that decrease the level of IgM significantly may also be beneficial. For example, proteasome inhibitors are highly active in WM and can decrease the IgM level.56,57 Bortezomib given IV twice a week induces neuropathy in some patients and therefore may not be the recommended agent of choice.47,56,57 However, the use of other proteasome inhibitors that do not induce neuropathy or, potentially, the use of subcutaneous bortezomib once a week may be useful in patients with IgM-related neuropathy. Another agent that has shown improvement of neuropathy in our clinic is the mTOR inhibitor everolimus (I.M.G., S. Treon, unpublished observation). This agent has shown 70% response rate as a single agent in patients with WM,58 and we have observed clinical benefit with neuropathy in many of these patients. Future studies are needed to examine the role of proteasome inhibitors or mTOR inhibitors in the treatment of IgM-related neuropathy.
Future directions and conclusion
WM is a distinct lymphoproliferative disorder characterized by IgM monoclonal secretion and lymphoplasmacytic cells in the BM. Although it is rare, significant advances have been made in our understanding of the pathogenesis and biology of this disease. Multiple novel agents have been developed for the treatment of WM and have shown good responses. In addition, agents such as bortezomib or everolimus show good responses in WM, but not specifically in other lymphomas or plasma cell dyscrasias, respectively. Therefore, accurate diagnosis and differentiating WM from other diseases is critical to adequately treating these patients and obtaining significant responses.
Acknowledgments
This study was supported in part by the International Waldenstrom Macroglobulinemia Foundation (1R01F0003743), Heje fellowship, and Kirsch Laboratory for Waldenstrom at Dana-Farber Cancer Institute, Boston, MA.
Disclosures
Conflict-of-interest disclosure: The author is on the board of directors or an advisory committee for Millennium, Onyx, and Novartis. Off-label drug use: None disclosed.
Correspondence
Irene M. Ghobrial, MD, Associate Professor of Medicine, Dana-Farber Cancer Institute, Harvard Medical School, 44 Binney St, D1B30, Boston, MA 02115; Phone: 617-632-4198; Fax: 617-582-8608; e-mail: Irene_ghobrial@dfci.harvard.edu.
