
Abstract
Leukocytosis, or elevated WBC count, is a commonly encountered laboratory finding. Distinguishing malignant from benign leukocytosis is a critical step in the care of a patient, which initiates a vastly different decision tree. Confirmation of the complete blood cell count and the WBC differential is the first step. Examination of the PB smear is essential to confirming the automated blood cell differential or affirming the manual differential performed on the PB smear. Next is separation of the leukocytosis into a myeloid versus a lymphoid process. Distinguishing a reactive lymphoid proliferation from a lymphoproliferative disorder requires examination of lymphocyte morphology for pleomorphic lymphocytes versus a monomorphic population, with the latter favoring a lymphoproliferative neoplasm. Samples suspicious for lymphoproliferative disorders can be confirmed and characterized by flow cytometry, with molecular studies initiated in select cases; precursor lymphoid neoplasms (lymphoblasts) should trigger a BM examination. Myeloid leukocytosis triggers a differential diagnosis of myeloid leukemoid reactions versus myeloid malignancies. The manual differential is key, along with correct enumeration of blasts and blast equivalents, immature granulocytes, basophils, and eosinophils and identifying dysplasia to identify myeloid malignancies. Confirmation and characterization of myeloid malignancies should be performed with a BM examination and the appropriate ancillary studies. Myeloid leukemoid reactions commonly result from infections and show activated neutrophil changes on morphology; these should prompt evaluation for infection. Other causes of reactive myeloid leukocytoses are also discussed herein.
Introduction
Leukocytosis is defined as an elevation of the WBC count for the patient's age. Using an appropriate reference interval is critical, because a WBC count of 30 × 109/L is considered elevated in an adult, but completely normal in the first few days of life. Similarly, the normal WBC differential also changes with age, with greater absolute numbers of neutrophils and lymphocytes seen in newborns and the normal “elevation” of lymphocytes persisting throughout childhood.1 Hospital laboratories are required to validate reference ranges in their respective patient populations as part of the validation process of automated hematology instruments.2
Automated differentials
Automated hematology analyzers rapidly analyze whole blood samples for the complete blood cell count and differential, including WBC count, RBC count, platelet count, hemoglobin concentration, hematocrit, RBC indices, and the leukocyte differential.3 The newer generation of hematology analyzers can examine thousands of leukocytes using flow cytometry–based methodology, some in combination with cytochemistry or fluorescence or conductivity, to elucidate different types of WBCs, including neutrophils, lymphocytes, monocytes, basophils, and eosinophils (ie, the 5-part differential). Nucleated RBCs are also detected routinely, and some analyzers also quantitate immature granulocytes.4 Based on proprietary algorithms, hematology analyzers will flag high or low numbers of specific cell types and possibly abnormal populations of leukocytes, including immature granulocytes, blasts, and variant/atypical lymphocytes. The automated hematology analyzers used in laboratories within physicians' offices may use older methodologies for cell analysis, such as impedance technology, which generates a 3-part leukocyte differential that includes lymphoid cells/basophils, neutrophils/eosinophils, and monocytes/other mononuclear cells. Whereas these smaller instruments may work well for screening a normal, healthy population, the presence of abnormal cells may cause error flags and inaccurate counts, resulting in a high review rate when used in a hospital-based population. Spurious elevations of the WBC count can also be seen, including platelet clumps, nucleated RBCs, incomplete lysis of RBCs, cryoglobulins, and cryofibrinogen.5 Regardless of the instrument used, a flagged blood sample triggers action before the CBC and WBC differential can be released; most commonly, this is a review of a peripheral blood (PB) smear. International groups have suggested criteria for action after automated CBC and WBC differential analysis.6
Examination of the PB smear
The PB smear review is a qualitative examination of the blood smear to detect clinically significant abnormalities in all cells, including leukocytes, erythrocytes, and platelets.7 A slide review is appropriate in all patients with unexplained leukocytosis, which requires proper slide preparation and staining. Blood smears are prepared from fresh, anticoagulated blood or from a drop of fresh blood from a syringe or finger-stick puncture. Blood films may be prepared manually using slides or coverslips or by instruments. A properly prepared smear is wedge shaped with a slightly rounded edge and shows a normal gradation of blood from thick to thin. The edges of the smear should not touch the edge of the slide because this area must be available for scanning. After air drying, the slides should be stained with Romanowsky-type stains such as May-Grünwald-Giemsa and Wright stains; DNA and RNA are labeled a deep basophilic color, the nuclei blue-purple, and the cytoplasm pink, depending on cytoplasmic granule composition. The area between cells should be clean and free of precipitate.
Beginning at low power, the slide should be examined to determine abnormalities in cell number, cell type, and aggregation. In manual prepared smears, larger WBCs tend to collect at the edges of the smear.8 Good practice for slide review requires assessment of leukocytes, erythrocytes, and platelets in both quantity and quality, correlating with automated cell counts. For RBCs, the blood film is examined in the area of the smear called the “feathered” edge, where the erythrocytes are barely overlapping or just touching. WBCs may be viewed in this area or in slightly thicker areas of the smear. In addition to number, WBCs should be examined for leukocyte composition and inclusions and for the presence of abnormal cells. This is performed at higher power usually using an oil-immersion lens. Whereas in many patients, a slide review may be adequate to confirm automated cell counts, in other cases (eg, abnormal cells, inaccurate automated leukocyte differential, or spurious leukocytosis), a manual leukocyte differential is necessary. Typically, 100 leukocytes are counted using microscopy. This low number of cells counted (compared with the thousands of leukocytes enumerated by automated analyzers) contributes to the high coefficient of variation in manual cell counts and highlights the problems with statistical sampling errors. Newer techniques for leukocyte classification include digital microscopy, in which computer algorithms classify cells and more leukocytes can be examined.9 Cell misidentification and unequal distribution of cells on a slide can contribute to errors. With regard to leukocytes specifically, neoplastic cells such as blasts and the lymphoid cells of lymphoproliferative disorders such as chronic lymphocytic leukemia (CLL) are extremely fragile and crush easily with mechanical pressure, resulting in increased numbers of “smudge” or “basket” cells. Leukocytes may also undergo apoptosis and other changes in morphology when aged samples are used, so smears must be prepared promptly from fresh specimens.
WBCs typically seen in a blood film include neutrophils, lymphocytes, monocytes, eosinophils, and basophils. Immature granulocytes (ie, blasts, promyelocytes, myelocytes, and metamyelocytes) and lymphoma cells are abnormal findings. Neutrophils include segmented and band forms. Bands constitute approximately 5%-10% of the nucleated cells in the blood under normal conditions; increased bands may be seen in certain pathologic states. However, with this method, the interobserver reproducibility of the band count is poor, rendering the band count of questionable value in most patients.10
Blast cells
Approaching leukocytosis in a patient requires confirmation of the leukocyte differential count to determine the types of cells present and which type is elevated.11 This is performed by examining a PB smear and either confirming the automated cell differential or performing a manual leukocyte differential. Distinguishing a myeloid process from a lymphoid process is essential. However, in some patients, examination of the smear may only show blast cells (Figure 1), and this is typically accompanied by anemia and thrombocytopenia. Morphologic features alone are unreliable in distinguishing blast type, so immunophenotypic studies are necessary to distinguish myeloid from lymphoid blasts. Such patients should have a BM examination with tissue obtained for aspirate smears, trephine biopsy, flow cytometric immunophenotyping, and cytogenetic and molecular genetic studies to classify acute leukemias/precursor neoplasms properly according to the World Health Organization classification.12 In some cases, cytochemical stains may also be useful. If an adequate aspirate smear is unable to be performed (a “dry tap”), touch preparations should be prepared from the fresh trephine biopsy and additional fresh BM cores should be sent for flow cytometric phenotyping and cytogenetic/molecular genetic studies. A sample containing blast cells requires careful examination for features suggesting a myeloid or lymphoid lineage. Auer rods or granules suggest a myeloid lineage, although lymphoblasts with granules may also be seen. Additional blast features such as large salmon-colored granules, prominent hofs, butterfly-shaped nuclei, and background dysplasia may suggest a certain type of acute myeloid leukemia. In particular, acute promyelocytic leukemia (APL) shows a characteristic morphology often accompanied by changes of microangiopathic hemolytic anemia (eg, schistocytes), because these patients may present with bleeding due to disseminated intravascular coagulopathy. The more common hypergranular variant of APL shows hypergranular blasts with many fine cytoplasmic granules and Auer rods; bundles of Auer rods may be seen in a small percentage of cells. The hypogranular variant of APL on morphology shows agranular blasts, but will show the typical nuclear features of APL with folded nuclei (Figure 1). Flow cytometry typically shows a characteristic phenotype with lack of expression of HLA-DR and usually a lack of CD34. Rapid FISH or RT-PCR testing for the PML-RARA fusion gene is typically performed. Other blast morphologies are shown in Figure 1.

Acute leukemia and precursor neoplasms. (A) Precursor B-lymphoblastic leukemia with a very high WBC with numerous blasts and smudge cells. (B) Precursor B-lymphoblastic leukemia with 2 large lymphoblasts. (C) Precursor T-lymphoblastic leukemia with a lymphoblast containing a moderate amount of basophilic cytoplasm. (D) APL with t(15;17)(q22;q12); PML-RARA, the hypogranular variant with butterfly-shaped nuclei. (E) APL with an abnormal promyelocyte containing multiple Auer rods below a lymphocyte. (F) Transient abnormal myelopoiesis associated with Down syndrome in an infant with numerous large blasts, hypogranular platelets, nucleated RBCs, a megakaryocytic nuclear fragment, and scattered schistocytes. (G) Acute monoblastic leukemia with abundant lightly granular cytoplasm containing vacuoles. (H) Acute myeloid leukemia with t(8;21)(q22;q22); RUNX1-RUNX1T1 with a small myeloblast containing a thin, delicate Auer rod. (I) Recurrent acute myeloid leukemia with associated microangiopathic hemolysis (schistocytes) and a hypogranular platelet. The 2 myeloblasts at bottom right contain partially condensed chromatin and sparsely granular cytoplasm, features sometimes found in blasts seen in myeloid neoplasms with myelodysplasia.
Acute leukemia and precursor neoplasms. (A) Precursor B-lymphoblastic leukemia with a very high WBC with numerous blasts and smudge cells. (B) Precursor B-lymphoblastic leukemia with 2 large lymphoblasts. (C) Precursor T-lymphoblastic leukemia with a lymphoblast containing a moderate amount of basophilic cytoplasm. (D) APL with t(15;17)(q22;q12); PML-RARA, the hypogranular variant with butterfly-shaped nuclei. (E) APL with an abnormal promyelocyte containing multiple Auer rods below a lymphocyte. (F) Transient abnormal myelopoiesis associated with Down syndrome in an infant with numerous large blasts, hypogranular platelets, nucleated RBCs, a megakaryocytic nuclear fragment, and scattered schistocytes. (G) Acute monoblastic leukemia with abundant lightly granular cytoplasm containing vacuoles. (H) Acute myeloid leukemia with t(8;21)(q22;q22); RUNX1-RUNX1T1 with a small myeloblast containing a thin, delicate Auer rod. (I) Recurrent acute myeloid leukemia with associated microangiopathic hemolysis (schistocytes) and a hypogranular platelet. The 2 myeloblasts at bottom right contain partially condensed chromatin and sparsely granular cytoplasm, features sometimes found in blasts seen in myeloid neoplasms with myelodysplasia.
Lymphoid leukocytosis
Reactive lymphocytosis
Distinguishing reactive lymphocytes from lymphoma cells can be challenging, but several key features should be kept in mind. First, the age of the patient helps to focus the differential diagnosis. Many of the lymphomas involving the PB are much more common in middle-aged to elderly adults than in children or infants. For example, certain lymphomas (ie, Burkitt lymphoma) are seen more commonly in children. Second, accurate clinical history can be critical, because any prior diagnosis of lymphoma can aid in the identification of these cells. Third, a well-prepared and stained PB smear is important. Old blood and poorly prepared samples can lead to misdiagnoses. When looking at the peripheral smear, a scan should be performed at 10× examining the overall slide before going to higher magnification. Reactive lymphocytoses will show a wide range of cellular sizes and shapes, with a lymphocytosis defined as an increase in the absolute lymphocyte count above that expected in an individual of the same age. Absolute lymphocyte counts are higher in children and infants compared with adults, so the appropriate reference intervals must be used. In adults, an absolute lymphocyte count of > 3.5 × 109/L can be considered lymphocytosis.
The classic example of reactive lymphocytosis is infectious mononucleosis (eg, EBV infection), in which a variety of lymphocytes are seen, ranging from small lymphocytes with round nuclei and condensed chromatin to reactive lymphocytes with an abundant pale blue cytoplasm, round to oval nuclei, and moderately condensed chromatin (Figure 2). The cytoplasm of these reactive lymphocytes may “hug” adjacent RBCs and show a basophilic rim at their margins. Immunoblasts are often present, seen as large lymphocytes with round to oval nuclei containing one or more prominent nucleoli. The cytoplasm of an immunoblast is abundant and deeply basophilic. Plasmacytoid lymphocytes may also be seen. Reactive lymphocytosis may be seen with several different viral infections, other infections, drug effects, stress, and secondary to malignancy (Table 1).

Lymphoid leukocytosis. (A) Infectious mononucleosis with a reactive lymphocytosis including an immunoblast at top. Note the pleomorphism of the lymphocytes. (B) A reactive lymphocytosis is seen in this patient with massive trauma due to a vehicular accident. (C) Large granular lymphocytosis, reactive. (D) CLL with characteristic small, round lymphocytes containing coarse, blocky chromatin. (E) Prolymphocytoid transformation in CLL. (F) Splenic marginal zone lymphoma with villous lymphocytes containing bipolar cytoplasmic projections. (G) Blastic MCL. (H) Lymphoplasmacytic lymphoma. (I) Circulating follicular lymphoma with clefted lymphoma cells.
Lymphoid leukocytosis. (A) Infectious mononucleosis with a reactive lymphocytosis including an immunoblast at top. Note the pleomorphism of the lymphocytes. (B) A reactive lymphocytosis is seen in this patient with massive trauma due to a vehicular accident. (C) Large granular lymphocytosis, reactive. (D) CLL with characteristic small, round lymphocytes containing coarse, blocky chromatin. (E) Prolymphocytoid transformation in CLL. (F) Splenic marginal zone lymphoma with villous lymphocytes containing bipolar cytoplasmic projections. (G) Blastic MCL. (H) Lymphoplasmacytic lymphoma. (I) Circulating follicular lymphoma with clefted lymphoma cells.

There are some exceptions to the rule of a pleomorphic lymphocytosis equating to reactive lymphocytosis. Infection by Bordetella pertussis gives a very characteristic reactive lymphocytosis composed of small, mature lymphocytes with deep nuclear clefts with a monomorphic appearance that can raise concern for lymphoma; however, clinical history will help with this diagnosis. Polyclonal B-lymphocytosis can also show lymphocytes with distinct nuclear clefts, but will demonstrate a spectrum of morphologic changes, including nuclear lobation and binucleate forms. This disorder is typically found in young to middle-aged women who smoke, have a high association with HLA-DR7, and show a longstanding lymphocytosis that is polyclonal by flow cytometry, but some genetic abnormalities have been documented.13 In addition, large granular lymphocytosis, an increased number of similar appearing large granular lymphocytes (a normal component of the blood), may be seen with viral infections, malignancy, after BM transplantation, and after chemotherapy (Figure 2). The persistence of a large granular lymphocytosis with accompanying neutropenia and variable anemia should trigger an evaluation for T-cell large granular lymphocytic leukemia (T-LGL), including flow cytometry and TCR gene rearrangements.14 Typical immunophenotypic findings of T-LGL include expression of CD3, CD8, CD57, and TCRα/β; TCR gene rearrangement is positive, which is helpful in excluding reactive large granular lymphocytes. Confirmatory testing for infections can be performed if the clinical history and physical examination are consistent with a specific type of infection; most commonly, this is a heterophilic Ab test (ie, a monospot test) for EBV infection.
Lymphoproliferative neoplasms
Whereas reactive lymphocytes are heterogenous, lymphoma cells tend to be homogenous. A PB smear may contain a subset of lymphoma cells amidst a background of normal lymphocytes, but those lymphoma cells will typically resemble one another. Lymphoma cells can exhibit a variety of morphologic appearances with various sizes and shapes, ranging from 8-30 μm in size and variable amounts of cytoplasm, depending on the type of lymphoma (Figure 2). The overall frequency of PB involvement by BM lymphoma was shown to be almost 30% in a study using a combination of morphology and flow cytometry, although it should be noted that this study specifically excluded chronic leukemias15 (Table 2).
Frequency of PB involvement by BM lymphoma

Adapted with permission from Arber and George.16
NOS indicates not otherwise specified; and NK, natural killer.
*8 of 9 cases of splenic marginal zone lymphoma showed PB involvement.
†A few cases with limited material were classified as high-grade, not otherwise specified and low-grade, not otherwise specified.
‡Not meeting the criteria for CLL.
Chronic lymphocytic leukemia, prolymphocytoid leukemia
CLL is the most common leukemia in adults in the Western world and therefore it is the most common neoplasm seen in the PB. Whether a patient has CLL or a monoclonal B-cell lymphocytosis (< 5 × 109/L of clonal B cells in a healthy person without symptoms, cytopenias, or lymphadenopathy), the morphology of the lymphocytes in the blood will be the same. Monoclonal B-cell lymphocytosis occurs in 3% of healthy persons, usually shows a CLL immunophenotype, and will progress to CLL in a small number of patients; it is important to be aware of the criteria for this clonal benign lymphocytosis to avoid overdiagnosis of lymphoproliferative disorders.16 Typical CLL morphology shows round, small nuclei with coarse blocky chromatin and frequent smudge cells; an albumin preparation may be prepared in the laboratory to prevent the formation of smudge cells. Atypical CLL morphology includes cleaved nuclei or larger lymphoid cells with slightly less condensed chromatin and those with a lower nuclear/cytoplasmic ratio. Up to 55% prolymphocytes may now be seen with CLL. Prolymphocytes are larger than normal lymphocytes, ranging from 10-18 μm in size, with a central oval to round nucleus, abundant blue cytoplasm, and, typically, a single prominent nucleolus. More than 55% prolymphocytes in the blood at initial diagnosis is seen in B-cell prolymphocytic leukemia (B-PLL) patients. Large cell transformation of CLL may show large cell lymphoma cells in the PB similar to those described below under diffuse large B-cell lymphoma. T-cell PLL (T-PLL) is similar to B-PLL in that both present primarily in elderly men with a striking leukocytosis and anemia and thrombocytopenia also commonly present. Morphology in T-PLL can range from lymphoid cells similar to the prolymphocytes of B-PLL to small lymphoid cells with cytoplasmic protrusions, occasional nuclear irregularities, condensed chromatin, and prominent nucleoli. Immunophenotyping shows a mature T-cell phenotype, including expression of CD7 and TCL1 expression by immunohistochemistry; a characteristic inversion (14)(q11q32) is present in the majority of cases as shown by cytogenetic studies.
Splenic marginal zone lymphoma, hairy cell leukemia
A high frequency of blood involvement by splenic marginal zone lymphoma has been described frequently.15,17 Splenomegaly, often with anemia or thrombocytopenia, is often accompanied by PB containing lymphoma cells with “villous lymphocytes” in which bipolar cytoplasmic projections are present. However, not all cases of splenic marginal zone lymphoma in the blood will contain lymphoma cells with prominent “villi.” These morphologic features often overlap with hairy cell leukemia. Hairy cells, in contrast, have spiky cytoplasmic projections extending from the entire periphery of the cell.
Mantle cell lymphoma
Similarly, a high frequency of PB involvement by mantle cell lymphoma (MCL) is now recognized.15,18 In MCL, lymphoid cells may range from small to intermediate-sized lymphoid cells with irregular nuclear contours to large blast-like cells resembling acute leukemia. Typical MCL cells are larger than lymphocytes with folded nuclei and a small amount of basophilic cytoplasm. Although nuclei are condensed, they contain more reticular chromatin and may contain a single prominent nucleolus, thus resembling prolymphocytes. Blastic or blastoid variants of MCL are the size of large cell lymphoma cells (20-30 μm) and have a moderate amount of basophilic cytoplasm; although nuclei are round to oval, they may be indented or convoluted. Chromatin is less condensed, resembling blastic chromatin, although these lymphoma cells are typically more variable than blasts seen in acute leukemia.
Burkitt lymphoma
Burkitt lymphoma cells are morphologically identical to the cells seen in Burkitt leukemia, with the distinction resting on whether there is less than or greater than 25% BM involvement, respectively. The Burkitt cell is moderate in size (10-25 μm) with an oval to round nucleus, moderately coarse chromatin, and 1-3 prominent nucleoli. A moderate amount of deeply basophilic cytoplasm is present and typically contains multiple small vacuoles. Surprisingly, a relatively high rate of diffuse large B-cell lymphoma may be seen in the blood, although this is likely underrecognized.19
Large cell lymphoma
Large cell lymphomas show some of the most abnormal morphology of the lymphomas. They may resemble a proliferation of immunoblasts with 1-2 prominent nucleoli or may present with deep convolutions with occasional nucleoli. These cells are large with small to moderate amounts of deeply basophilic cytoplasm, are occasionally vacuolated, and often show angulated and folded nuclear contours. Although these cells can be confused with blasts, large cell lymphoma cells vary in size more than blasts and lack the smooth, even chromatin found in blasts. “Double-hit” B-cell lymphomas, defined as B-cell lymphomas with break points involving MYC in combination with another recurrent break point (typically BCL2), show features of diffuse large B-cell lymphoma and Burkitt lymphoma and are associated with higher-stage disease, including frequent BM involvement.20
Follicular lymphoma
Follicular lymphoma, when it involves the blood, shows a characteristic morphology. These lymphoma cells are slightly bigger than normal lymphocytes with a cleft appearance and moderately coarse chromatin; occasionally, large neoplastic cells may also be seen. Cytoplasm is typically scant, but may be moderate, and the majority of nuclei show folds and convolutions. Nucleoli may be present.
Other types of lymphoma cells also show angulated nuclei, including MCL, Sézary syndrome, and adult T-cell leukemia/lymphoma (ATLL). MCL cells lack the coarse chromatin of follicular lymphoma and are usually larger in size with gentle nuclear folds, not the deep clefts of follicular lymphoma. Sézary cells, characteristic of Sézary syndrome, show convoluted nuclear membranes with a cerebriform appearance and contain dark, hyperchromatic chromatin typically lacking nucleoli. Patients with Sézary syndrome have a generalized erythroderma with blood involvement by malignant T cells typically expressing CD4 and other pan-T-cell antigens with absent or diminished expression of CD7. The malignant T cells of ATLL are present in high number in the PB, with prominent nuclear irregularities and nuclear lobations; other cytopenias may also be present. ATLL is an aggressive disease caused by HTLV-1, and is more commonly seen in Japan, Africa, the Caribbean, and the southeastern United States. The immunophenotype is that of a mature T-cell lymphoma with expression of CD2, CD3, CD4, CD5, and CD25 with diminished to absent expression of CD7. Serologic or molecular studies can confirm HTLV-1 infection when ATLL is suspected. Molecular genetic studies can be helpful in the differential diagnosis of cleaved lymphocytes. FISH for CCND1/IGH@ is useful for confirmation of circulating MCL. FISH or RT-PCR can detect the BCL2-IGH@ rearrangement representing the t(14;18)(q32;q21) of follicular lymphoma. However, the BCL2-IGH@ rearrangement has also been reported in the blood of healthy donors.21 The malignant T cells of Sézary syndrome show TCR gene rearrangements; T-cell gene rearrangements are also found in other T-cell lymphomas, including ATLL and T-LGL. In lymphoplasmacytic lymphoma, plasmacytoid lymphocytes, small to intermediate-sized lymphocytes, and even frank plasma cells may be seen. The distinction among the lymphoma types requires flow cytometric immunophenotyping, as shown in Table 3 for the small B-cell lymphoproliferative disorders.
Common marker expression patterns in small B-cell lymphoproliferative disorders involving the blood

Adapted with permission from Pereira et al,7 page 153.
LPL indicates lymphoplasmacytic lymphoma; SMZL, splenic marginal zone lymphoma; HCL, hairy cell leukemia; FL, follicular lymphoma; sIg, surface immunoglobulin; and cIg, cytoplasmic immunoglobulin.
Myeloid leukocytosis
Myeloid leukocytosis may represent granulocytosis (ie, neutrophilia, eosinophilia, and basophilia) or monocytosis. Neutrophilia is defined as an elevated circulating neutrophil count (> 7.7 × 109/L in adults). The absolute neutrophil count can be determined by multiplying the total WBC count by the percentage of polymorphonuclear cells (segmented neutrophils) and band forms. Reactive neutrophilia (neutrophilic leukemoid reaction) results from an increased production of neutrophils, demargination (process of neutrophils entering the peripheral circulation from areas of intravascular marginated neutrophil pools), or a decreased egress of neutrophils from the peripheral circulation to the tissues. Neutrophilic leukemoid reactions are seen commonly with infections (bacterial, viral, mycobacterial,22 or treponemal), but any strong stimulus to the BM can trigger this reaction. Causes of neutrophilia include malignancy, inflammation, drugs (eg, glucocorticoids or lithium), myeloid growth factors, hemorrhage, and splenectomy.23–29 Neutrophilic leukemoid reactions show morphologic overlap with chronic myelogenous leukemia (CML), acute myeloid leukemia, chronic neutrophilic leukemia, and myelodysplastic/myeloproliferative neoplasms. Whereas a neutrophilic leukemoid reaction rarely has a WBC count > 50 × 109/L, CML may far exceed that value; however, a lower WBC does not preclude a consideration of CML. Immature granulocytes (ie, promyelocytes, myelocytes, and metamyelocytes), which are indicative of a left shift in the PB, may be seen with either reactive neutrophilias or myeloid neoplasms such as CML. A more pronounced left shift is typical of CML accompanied by absolute increases in the numbers of basophils and eosinophils, but a leukemoid reaction must be excluded (Figure 3). FISH or molecular studies for BCL-ABL1 can be performed in PB to confirm the diagnosis of CML, although BM examination is recommended in new diagnoses with accompanying metaphase-based cytogenetic studies. Activated neutrophils (eg, toxic granulation, Dohle bodies, and cytoplasmic vacuoles) are more characteristic of a reactive neutrophilia such as that seen in patients with bacterial infections or those receiving growth factors; cytoplasmic vacuoles alone must be interpreted with caution because this can also be seen in old samples. Other features favoring a reactive neutrophilia include circulating fragments of neutrophil cytoplasm, thrombocytosis, and a lack of basophilia; eosinophilia and monocytosis may also be present. If the left shift is accompanied by nucleated RBCs, a leukoerythroblastic smear may suggest a BM-infiltrative process such as tumor or fibrosis. In the latter process, this is typically accompanied by increased numbers of dacrocytes (teardrop-shaped cells). As mentioned previously, increased numbers of circulating blasts should prompt a BM examination for acute leukemia. Leukoerythroblastic smears can also be seen in patients receiving growth factors.

Myeloid leukocytosis. (A) Neutrophilia in a patient with a perforated tumor and infection. (B) G-CSF effect with immature granulocytes. (C) Neutrophils with toxic granulation and vacuoles are seen in a patient with a bacterial infection. (D) CML, BCR-ABL1+, chronic phase, shows many immature granulocytes and occasional blasts. (E) CMML with abnormal monocytes and a large hypogranular platelet (adapted with permission from Pereira et al,7 page 101). (F) Myeloid neoplasm with eosinophilia and PDGFRA shows 2 eosinophils with eosinophilic granules that do not completely fill the cytoplasm and occasional hypogranular platelets.
Myeloid leukocytosis. (A) Neutrophilia in a patient with a perforated tumor and infection. (B) G-CSF effect with immature granulocytes. (C) Neutrophils with toxic granulation and vacuoles are seen in a patient with a bacterial infection. (D) CML, BCR-ABL1+, chronic phase, shows many immature granulocytes and occasional blasts. (E) CMML with abnormal monocytes and a large hypogranular platelet (adapted with permission from Pereira et al,7 page 101). (F) Myeloid neoplasm with eosinophilia and PDGFRA shows 2 eosinophils with eosinophilic granules that do not completely fill the cytoplasm and occasional hypogranular platelets.
Eosinophilia
Most eosinophilias, typically defined as > 1.5 × 109/L eosinophils, are reactive in nature and should be investigated for a reactive cause of eosinophilia; these include: infections (especially parasites), allergy/hypersensitivity diseases, connective tissue diseases, pulmonary diseases, cardiac diseases, dermatologic disorders, malignancy (eg, lymphomas including Hodgkin lymphoma and carcinomas), gastrointestinal disease, and adrenal insufficiency.30 Once a reactive cause is excluded, investigations into primary eosinophilias should be performed, including BM examination, cytogenetic studies, and FISH or PCR studies for the FIP1L1-PDGFRA fusion gene (this is a cryptic deletion not seen by standard cytogenetic karyotyping) (Figure 3).30 Cytogenetic studies will detect abnormalities indicative of PDGFRB and FGFR1. Immunohistochemistry can be performed on the trephine biopsy to identify mast cell proliferations that may be seen in those myeloid neoplasms with eosinophilia. Those myeloid neoplasms with eosinophilia associated with PDGFRA mutations are sensitive to imatinib mesylate. Both hypereosinophilic syndrome and chronic eosinophilic leukemia, not otherwise specified will have an eosinophilia ≥ 1.5 × 109/L; clonality or increased blasts indicates chronic eosinophilic leukemia once the myeloid neoplasms with eosinophilia and PDGFRA, PDGFRB, and FGFR1 are excluded. An absence of clonality, no increase in blasts, no evidence for a reactive cause of eosinophilia, and a persistent eosinophilia for at least 6 months with organ involvement and dysfunction are diagnostic of hypereosinophilic syndrome.30 T-cell phenotyping and T-cell clonality studies are also recommended in the workup of eosinophilia to identify the lymphocyte-variant hypereosinophilia.30
Basophilia
Basophilia, defined as > 0.3 × 109/L in adults,11 is extremely uncommon and, if confirmed, should prompt a BM examination given its strong association with myeloproliferative neoplasms (although rare reactive basophilias have been reported). Distinguishing basophils from mast cells may also be difficult.
Monocytosis
An absolute monocytosis is defined as > 1 × 109/L monocytes. A reactive monocytosis may be seen with malignancy (ie, carcinoma, plasma cell myeloma, or lymphoma), chronic infections, autoimmune disorders, and splenectomy. It is also seen in a regenerating BM such as after BM transplantation or chemotherapy. The persistence of an absolute monocytosis should prompt examination of the BM with flow cytometric immunophenotyping and cytogenetic studies, because persistent monocytosis raises a differential diagnosis, including chronic myelomonocytic leukemia (CMML), acute monoblastic/monocytic leukemia, CML, juvenile myelomonocytic leukemia, atypical (BCR-ABL negative) CML, and myelodysplastic/myeloproliferative neoplasms, unclassifiable.33 Molecular genetic testing, although not now routinely available, may be used in the future to help distinguish the various MPNs/MDSs (eg, PTPN11, RAS, NF1, TET2, and RUNX1). It is not uncommon for patients with acute monoblastic/monocytic leukemia to present with more differentiated monocytes in the PB, so such cases of “CMML” should always be confirmed with BM examination. CML, CMML, and atypical CML can be distinguished as shown in Table 4 (Figure 3).
PB examination in the differential diagnosis of CMML, CML, and atypical CML
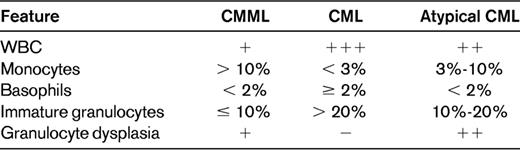
The WBC count is higher in CML than either CMML or atypical CML, including WBCs greater than 200 × 109/L. The percentage of monocytes is generally greater than 10% in CMML, higher than in either CML or atypical CML. CML shows a prominent basophilia compared with CMML and atypical CML. Large numbers of immature granulocytes are typical of CML, with fewer numbers in atypical CML and even less in CMML. Greater numbers of circulating blasts are seen in atypical CML than in either CMML or chronic-phase CML. Finally, atypical CML has the most granulocytic dysplasia, which is generally absent in CML and may be present or absent in CMML.
Adapted with permission from Pereira et al,7 page 99.
In conclusion, leukocytosis in a patient should prompt confirmation of the CBC and WBC differential. Examination of the blood smear should be performed to establish a manual differential or to confirm the automated differential. This will allow the distinction of myeloid from lymphoid disorders. Distinguishing myeloid leukemoid reactions from myeloid malignancies is difficult, with features such as dysplasia, basophilia, WBC count > 50 × 109/L, a pronounced left shift, and increased blasts favoring a myeloid malignancy with recommended BM examination and appropriate ancillary testing. Myeloid leukemoid reactions may be seen with a variety of stimuli, but markers of infection such as activated neutrophils can be helpful features to look for in conjunction with the appropriate laboratory testing. With respect to lymphocytoses, pleomorphic lymphocytosis in the appropriate clinical context favors a reactive lymphocytosis, whereas a homogenous population of lymphoid cells favors a lymphoproliferative disorder. Flow cytometric immunophenotyping is typically recommended for further classification of lymphoproliferative disorders, with molecular genetic testing as appropriate. Regardless of whether a myeloid or lymphoid leukocytosis is favored, the presence of increased blasts should prompt BM examination with appropriate ancillary studies. An algorithmic approach as shown in Figure 4 may be helpful. Specific details for evaluation of eosinophilia are best addressed by Gotlib.30

Diagnostic algorithm for the workup of leukocytosis. If increased blasts are present, then evaluation for an acute leukemia or precursor neoplasm should begin with BM examination including appropriate ancillary studies. If myeloid cells are present, the leukocytosis should be stratified into neutrophilia, monocytosis, basophilia, or eosinophilia; more than one type of leukocytosis may be present. Most monocytoses are reactive in nature. However, if reactive causes have been excluded, a persistent monocytosis of more than 3 months or the findings of dysplasia, blast cells, or significant left shift should trigger a BM examination to evaluate for malignancy. Neutrophilia should prompt examination for left shift, signs of activated neutrophils, basophilia, dysplasia, and degree of leukocytosis. Most neutrophilias are reactive in nature. A WBC count < 50 × 109/L, but usually < 30 × 109/L, is typical. Signs of activated neutrophils, mild left shift, and an absence of basophilia all suggest a reactive process. A marked leukocytosis of > 50 × 109/L, marked left shift, dysplasia, or basophilia should prompt a BM examination to evaluate for a myeloid malignancy. Basophilia, although rare, is most suggestive of a MPN, especially CML. PCR for BCR-ABL1 and JAK2 mutational studies can be performed in blood, but a BM examination with cytogenetic studies should also be performed. Most eosinophilias are reactive in nature and these should be evaluated as outlined by Gotlib.30 Once reactive eosinophilias are excluded, myeloid and lymphoid neoplasms with eosinophilia and PDGFRA, PDGFRB, and FGFR1 should be searched for by performing a BM examination, cytogenetic studies, and FISH or PCR for the PDGFRA mutation. If a lymphocytosis is present, the lymphoid cells should be examined for pleomorphism or monomorphism. A pleomorphic lymphocytosis favors a reactive lymphocytosis. Correlation with clinical findings is necessary; a monospot test for EBV or viral serologies can also be performed. If monomorphic lymphocytosis is present, a lymphoproliferative disorder should be searched for using flow cytometric immunophenotyping. Depending on these results, select molecular genetic tests will be helpful. A BM biopsy or extramedullary tissue biopsy may be necessary for a final diagnosis of lymphoma. CBC indicates complete blood cell count; MPN, myeloproliferative neoplasm; MPN eos, myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1; and FL, follicular lymphoma.
Diagnostic algorithm for the workup of leukocytosis. If increased blasts are present, then evaluation for an acute leukemia or precursor neoplasm should begin with BM examination including appropriate ancillary studies. If myeloid cells are present, the leukocytosis should be stratified into neutrophilia, monocytosis, basophilia, or eosinophilia; more than one type of leukocytosis may be present. Most monocytoses are reactive in nature. However, if reactive causes have been excluded, a persistent monocytosis of more than 3 months or the findings of dysplasia, blast cells, or significant left shift should trigger a BM examination to evaluate for malignancy. Neutrophilia should prompt examination for left shift, signs of activated neutrophils, basophilia, dysplasia, and degree of leukocytosis. Most neutrophilias are reactive in nature. A WBC count < 50 × 109/L, but usually < 30 × 109/L, is typical. Signs of activated neutrophils, mild left shift, and an absence of basophilia all suggest a reactive process. A marked leukocytosis of > 50 × 109/L, marked left shift, dysplasia, or basophilia should prompt a BM examination to evaluate for a myeloid malignancy. Basophilia, although rare, is most suggestive of a MPN, especially CML. PCR for BCR-ABL1 and JAK2 mutational studies can be performed in blood, but a BM examination with cytogenetic studies should also be performed. Most eosinophilias are reactive in nature and these should be evaluated as outlined by Gotlib.30 Once reactive eosinophilias are excluded, myeloid and lymphoid neoplasms with eosinophilia and PDGFRA, PDGFRB, and FGFR1 should be searched for by performing a BM examination, cytogenetic studies, and FISH or PCR for the PDGFRA mutation. If a lymphocytosis is present, the lymphoid cells should be examined for pleomorphism or monomorphism. A pleomorphic lymphocytosis favors a reactive lymphocytosis. Correlation with clinical findings is necessary; a monospot test for EBV or viral serologies can also be performed. If monomorphic lymphocytosis is present, a lymphoproliferative disorder should be searched for using flow cytometric immunophenotyping. Depending on these results, select molecular genetic tests will be helpful. A BM biopsy or extramedullary tissue biopsy may be necessary for a final diagnosis of lymphoma. CBC indicates complete blood cell count; MPN, myeloproliferative neoplasm; MPN eos, myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1; and FL, follicular lymphoma.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Tracy I. George, MD, Assistant Professor of Pathology, Director, Clinical Hematology Laboratory, Stanford University School of Medicine, 300 Pasteur Dr, Rm H1501B, Stanford, CA 94305-5627; Phone: 650-724-3356; Fax: 650-736-1473; e-mail: tigeorge@stanford.edu.
