
Abstract
For the past 5 decades, the care for hemophilia patients has improved significantly to the point that a newborn with hemophilia living in a developed nation can expect to have a normal lifespan and a high quality of life. Despite this, there are several new challenges that the hemophilia community will face in the coming years. First, the hemophilia community will soon be challenged with adopting a variety of new agents into clinical practice. Second, the normalization of patients' lives as a result of improved treatment has led to new problem areas, including obese/overweight hemophiliacs and osteoporosis. In addition, although mortality rates are similar to those of the healthy population, morbidities such as hemophilic arthropathy still occur. Third, the cost of care continues to rise, both due to the development of expensive new therapies and to the costs of managing problems such as obesity and osteoporosis. Finally, most patients in the world with hemophilia receive little to no care and although this is an enormous challenge, it must be confronted. This review discusses some new challenges facing developing nations and their care for hemophilia patients. In summary, in hemophilia in the coming few years, several new challenges will need to be confronted.
Introduction
Over the past 50 years, tremendous strides have been in made in the management of hemophilia such that a young boy born in a developed nation can expect to lead a nearly normal life free of the debilitating joint damage that is the hallmark of this disorder. As a result of this progress, new challenges have emerged and will continue to emerge, which will require both more research and more health care resources. First, whereas in the recent past, the main focus of innovation in hemophilia was the development of safer and more efficacious factor replacement therapies, the focus now is shifting toward the development of novel therapeutics with improved properties and to curative approaches such as gene therapy. Second, as patients' lives become more normal, they are developing problems that previously were either unknown and therefore ignored (eg, osteopenia) or did not exist (eg, obesity and aging issues). Third, as a result of the above 2 challenges, the cost of care for hemophilia continues to rise. Along with the expected high cost of new agents, the costs associated with managing new complications and an aging population will create new challenges in a world with more patients and fewer financial resources devoted to health care. Finally, no review of hemophilia challenges would be complete if it ignored the fact that the majority of patients throughout the world receive little to no treatment. Improving access to care and the quality of care in developing nations is a vital yet often ignored challenge. In the previous 2 years, the American Society of Hematology Education Program has had sessions on hemophilia dedicated to issues of aging (2010),1 the biological basis for novel therapeutics (2010 and 2011),2,3 and cost (2011).4 In addition, this year, gene therapy will be discussed on its own.5 Therefore, this review addresses the following challenges: reducing mortality and morbidity in the developed world, recognizing emerging threats such as obesity and osteopenia, understanding the potential problems associated with novel therapeutics, and assessing specific issues related to the management of hemophilia in the developing world.
Mortality and morbidity in the developed world
Mortality
Over the past century, the life expectancy of persons with hemophilia (PWH) has improved markedly to the point where some studies demonstrate a near normal life expectancy even for severe hemophiliacs.6–10 In the first half of the 20th century, several studies explored the life expectancy of PWH and demonstrated a significantly shortened life expectancy ranging from 16-23 years.11 In the latter half of the 20th century, life expectancy improved markedly to approximately 65 years.12 In the late 1970s and early 1980s, many patients, including the majority of severe hemophiliacs in countries with access to factor concentrates, contracted HIV and the hepatitis C virus (HCV) from infected plasma-derived products. This had a devastating effect on the hemophilia community, with many PWH dying from these infections.13 This led to a decreased life expectancy for a cohort of at risk patients: those born before approximately 1985 for HIV and approximately 1990 for HCV.6,7 More recently, studies examining life expectancy in hemophiliacs have controlled for the presence of HIV and HCV and, in PWH who are not infected, life expectancy is comparable to the general population.6,7 Among patients with inhibitors, only one study has examined mortality rates with respect to inhibitor development. Although mortality rates were higher in earlier cohorts of patients with inhibitors, in the most recent cohort studied (1993-1999), the mortality rates between severe hemophiliacs with and without inhibitors was the same.7 Therefore, further reducing mortality in patients who are not infected with HIV and HCV in the developing world may not be possible. This highlights the importance of the availability of factor concentrates that pose no threat for the acquisition of infectious agents.
Morbidity
This section explores the issues of bleeding-related morbidity with a focus on musculoskeletal disease and intracranial hemorrhage (ICH; Table 1). The main target site for bleeding in hemophilia is the joints. Before the widespread availability of clotting factor concentrates, PWH frequently developed crippling joint disease referred to as hemophilic arthropathy. With the implementation of prophylactic administration of factor concentrates, which was pioneered in Sweden more than 40 years ago, hemophilic arthropathy for most patients can be prevented completely.14 Over the past 20 years, prophylactic infusions of clotting factor concentrates has become the standard of care for patients with severe hemophilia in countries with essentially unlimited access to factor (a discussion of the challenges of hemophilia in the developing world is below). Recently, a randomized clinical trial demonstrated the protective effects of prophylaxis on joints,15 resulting in a Cochrane review recommending prophylaxis as the best practice for patients with severe hemophilia.16

Despite all of these advances, hemophilic arthropathy still occurs. First, for patients in whom neutralizing Abs (inhibitors) develop, existing therapies are not as effective at bleeding prevention as is standard replacement therapy.17 Second, patients with mild and moderate hemophilia, in whom serious bleeding episodes are less common and therefore do not usually receive prophylactic therapy, can also develop arthropathy as a result of poor recognition and/or undertreatment of bleeding episodes.18 Third, the penetration of the use of prophylactic therapy is not 100%, even in situations where the availability of clotting factor concentrates is not limited.19–21
Among the above 3 groups, PWH with inhibitors (hereafter referred to as inhibitor patients) suffer from the highest incidence and severity of joint disease.17 The currently available hemostatic agents available for bleed management, anti-inhibitor coagulation concentrate (FEIBA VH; Baxter) and recombinant factor VIIa (rFVIIa; Novoseven RT; Novo Nordisk), although efficacious, do not fully restore normal coagulation. Therefore, bleeding episodes in this group of patients are more difficult to treat, resulting in a more prolonged exposure of blood to the synovial lining of the joints and leading to synovitis, which eventually results in arthropathy. Until recently, the use of prophylaxis in inhibitor patients was not considered for a variety of reasons, chiefly the inability of the current products to fully restore normal coagulation and their relatively short half-lives compared with replacement therapies. Despite these shortcomings of FEIBA and rFVIIa, clinical trials using either of these agents in a prophylactic approach demonstrated a reduction of bleeding episodes in the 50% range overall; however, for some patients on these trials, bleeding reduction was in the 90% range.22,23 Therefore, prophylaxis could be considered as a potentially effective therapy in inhibitor patients, and an approach suggesting when to use prophylaxis in inhibitor patients has been published recently.24 Whether such prophylaxis in inhibitor patients reduces joint-related morbidity long-term, however, is unclear. The most effective way to reduce morbidity in inhibitor patients is to prevent inhibitors from developing in the first place. A better understanding of the mechanisms underlying inhibitor development, along with strategies aimed at minimizing risk factors that can be controlled, represent an important challenge for the coming years. Once an inhibitor develops, eradication becomes the chief goal, and immunotolerance therapy is the most effective approach. Unfortunately, this treatment is not effective in approximately 30% of patients.25 Therefore, another goal is to develop novel treatments that can eradicate inhibitors in all patients who develop them. Finally, developing agents that can better treat and even prevent bleeds in inhibitor patients remains an important goal, because both prevention of inhibitors and improved eradication strategies remain elusive.
For patients with mild and moderate hemophilia, and particularly for those who bleed rarely, the mere recognition that a bleed is occurring can be missed. Because bleeds in such patients are most often associated with trauma, the pain and swelling may be attributed to the trauma and not a hemophilic bleed. With the more widespread use of prophylaxis in severe hemophiliacs preventing most bleeding episodes, joint disease in moderate and mild hemophiliacs is increasingly being recognized.18 This potential irony of more joint disease in hemophiliacs with less severe disease is an important new challenge facing hemophilia care providers.
Lastly, despite the clear evidence for the benefits of prophylaxis in severe hemophiliacs, many patients still are not prescribed this regimen and even in those for whom it is prescribed, not all adhere to the recommendations.26,27 Recent surveys suggest that even for severe factor VIII–deficient patients, in whom the evidence for the benefits of prophylaxis are the most clear, the highest rate of prophylaxis is in children in Canada at 84%.20 Rates in other patients (eg, adults and those with factor IX deficiency) in Canada and in other countries are lower.20,21,27 The reasons for the incomplete penetration of prophylaxis are unclear; however, although some of these patients may not bleed enough to warrant prophylaxis, it is likely that the majority of these remaining patients simply are not receiving the standard of care. Even if prophylaxis is prescribed, this regimen may not be adhered to. Unfortunately, there are no firm data on the rate of adherence in hemophilia. Although a validated tool had been developed (VERITAS-Pro), it has not been adopted widely and there have been no studies reporting results using this tool since the validation study was published.28 Therefore, there are only estimates of adherence based on survey studies.27 Studies from several countries have had fairly consistent estimates of adherence for younger children (< 12 years) of approximately 85%, but a wider variance for teenagers and adults ranging from 18%-40%, which in either case is far lower than that for young children.29–31 Therefore, another future challenge is first to develop better estimates of adherence and then to determine scientifically the barriers to adherence. The major barriers likely include the pain and difficulty associated with venipuncture (and the frequent need in children for a venous access device), the frequency of the infusions, the time it takes to prepare for and administer the infusion even in those with good venous access, and the lack of perceived benefit.26 With a clearer view on the incidence of lack of adherence combined with more precise knowledge regarding the barriers, specific strategies to overcome the barriers can be developed.
A less common albeit potentially severely morbid event is ICH. Estimates on the incidence of ICH in the neonatal period average approximately 4%, whereas the data beyond the neonatal period are more variable and harder to interpret; the most recent studies suggest an incidence of approximately 2%-2.5%.32–35 Due to the rarity of this event, performing studies to determine the most appropriate therapy are not possible. In general, such a potentially devastating event warrants the initiation of secondary prophylactic therapy indefinitely for patients without inhibitors, whereas for those with inhibitors, expert guidelines suggest treatment for a minimum of 6 months.36 Due to the life-threatening nature of such events, the best therapy is prevention.
Although preventing neonatal ICH is not possible in families with no history of hemophilia, for those in which the mother is known to be a carrier, there are several strategies that could be considered to reduce or even eliminate the risk. The first would of course be to prevent the birth of hemophilia patients, which could technically be achieved via preimplantation genetic diagnosis. This issue clearly carries with it significant ethical issues that are beyond the scope of this review, but have been explored previously.37 Another controversial approach is to perform prenatal (postconception or postimplantation) genetic testing that could identify fetuses with hemophilia, allowing parents the option to terminate the pregnancy. Less controversial approaches include noninvasive sexing via ultrasound, which allows for the identification of male fetuses who could then be more carefully approached perinatally (see discussion in the next section on mode of delivery).
Mode of delivery (even assuming the fetus is an affected male) remains controversial. A recent series of publications has explored these issues in depth, most recently in debate format in the journal Haemophilia, and persuasive arguments are made both on the side of vaginal delivery and elective Caesarean section.38–40 Lastly, for patients outside of the perinatal/neonatal period, an added benefit of prophylaxis for the prevention of joint bleeding is the prevention of ICH.
Emerging threats
Obesity
The developing world is in the midst of an overweight and obesity epidemic, and rates of overweight and obese hemophiliacs roughly parallel those of nonhemophiliacs.41 This is not particularly surprising, because PWH are generally in good health and do not suffer from some of the problems found in more serious chronic disorders in which the metabolic rate might be elevated. Furthermore, some parents are highly protective of their hemophilic children and do not allow them to participate in physical activity, placing them at increased risk for becoming overweight. Although overweight and obese hemophiliacs are at risk for the same chronic problems as the general population, PWH are also at risk for additional problems. For example, it has been demonstrated that hemophilic arthropathy occurs more commonly in overweight and obese hemophiliacs, with a corresponding decrease in joint range of motion that is correlated with an increased body mass index.41 The precise mechanism by which joint disease is increased in overweight and obese PWH is not clear. Another issue that has not yet been clarified with respect to overweight and obese PWH is clotting factor concentrate dosing. Factor dosing is weight based, so heavier patients receive higher doses of factor. Unfortunately, it is not clear whether this is necessary because fatty tissue has less blood supply than muscle and lean tissues.42 Although dosing that is based on ideal body weight can have a substantial economic impact,43 this is neither the current practice nor is it clear that it should be. Considering that roughly 50% of patients are overweight or obese, clarifying how factor should be dosed in such patients should be the subject of additional research, with results that could potentially have a significant economic impact. Of course, aside from the economic costs are the health consequences for PWH who do not maintain a healthy weight.
Osteopenia
Although osteoporosis was first reported in PWH as far back as 1994,44 only recently has it become a focus for concern and research, most likely due to the lack of research and to the fact that, unlike bleeding symptoms, osteopenia is an “invisible” problem. In 1994, Gallacher et al first reported the association between hemophilia and osteoporosis.44 In that study, 19 male patients with severe hemophilia were compared with 19 age- and sex-matched controls, with the results demonstrating significantly lower bone mineral density (BMD) in the lumbar spine and femoral neck. More recently, a pediatric study confirmed the findings of the earlier study in adults, demonstrating that children with hemophilia are at increased risk for reduced BMD.45 Additional evidence can be gleaned from studies of bone biomarkers in PWH, which demonstrate that those with low BMD have reduced serum osteocalcin, a bone formation biomarker.46 Although the evidence for this association appears to be clear, the reasons for it have not been fully elucidated. The investigators of the above studies speculate that the link between hemophilia and osteopenia is the reduced physical activity experienced by patients who either cannot participate in physical activity due to arthropathy or do not participate as a result of overly protective parents or medical providers. One study attempted to correlate physical activity in adult hemophiliacs with BMD, but was only able to demonstrate a correlation between vigorous activity and BMD in the lumbar spine.47 Because this study was conducted in adults, all of whom received prophylactic factor infusions, it is possible that a clear effect of physical exercise could not be demonstrated in this high-functioning group. Further studies on this association in children and adolescents and studies comparing patients with more physical limitations may yet demonstrate that physical activity is indeed the link between PWH who do or do not have osteopenia. It should be noted that peak BMD is achieved by accruing bone during childhood and adolescence; therefore, although the symptoms of osteoporosis usually do not manifest until adulthood, preventive measures are best achieved during childhood. Currently, those caring for PWH should be aware that loss of BMD and subsequent osteoporosis are potential complications of hemophilia and that particular attention should be paid to those with more severe physical limitations. Although it is premature to recommend routine BMD testing for all PWH, selected patients may benefit from screening and subsequent therapeutic measures to correct the loss of BMD.
Challenges with novel therapeutics
In the near future, several new therapeutic agents with enhanced properties (ie, longer duration of action, increased potency, novel mechanisms of action) will become available. Although none of these agents is yet available, 2 recent reviews describing in detail the specific agents in development have been published in the ASH Education Book in 2010 and 2011.2,3 The present review does not discuss the specifics of these products, but rather speculates about the following potential challenges that may arise upon the approval of these new products: agent selection, laboratory monitoring, combining new agents with other new agents or with older agents, unforeseen adverse events, and cost (Table 2).
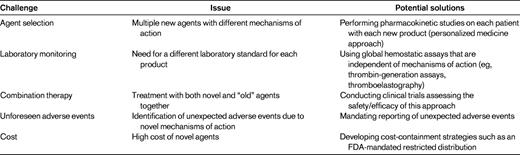
With respect to novel agent selection, it is likely that several agents with the same indications (prophylaxis for FVIII or FIX deficiency) will be available. How will a clinician select one versus another? Complicating this question is that the mechanisms for prolonging the half-life vary between the products in development (eg, PEGylation, Fc fusion, albumin fusion, and polysialylation). Although it is easy to argue that all of the recombinant FVIII products are essentially identical when it comes to efficacy and safety, one could speculate that with the novel agents some patients may have better outcomes with one type of product versus another because their half-life–altering mechanisms of action are different. For example, could some patients have more rapid clearance of PEGylated moieties than others while simultaneously having slower mechanisms for clearing Fc fusion products? If so, how could a clinician assess this? This issue will not be answered in the clinical trials leading to the approval of these agents. There are likely to be several other issues that will become apparent after approval of these medications (including cost, which is discussed below) that may affect agent selection when a choice is available.
Although laboratory monitoring of current factor replacement products is relatively straightforward, this will not remain the case as new products are introduced. How one will measure FVIII or FIX activity in patients receiving novel agents is unclear. One possibility is that for each product, a specific reference standard will be required. Such a scenario will significantly complicate matters for both the clinician and the clinical laboratory, because the clinician will need to report to the laboratory which product a patient is on and the laboratory will need to set up standard curves using multiple references; this is fraught with problems with a high potential for error. Taking this one step further, will a given factor level reported by the laboratory be comparable to a factor level for the currently available products? In other words, does a level of 20% for a PEGylated or Fc fusion molecule mean the same thing and does that equate to a 20% level for someone who receives a current version of rFVIII? Again, there are several other such questions/scenarios that will represent new challenges for the care of PWH.
Yet another challenge will involve the use of both novel long-acting agents with the currently available agents. For example, although an extended half-life product will no doubt improve the quality of life for patients, it is important to realize that a once-weekly product that can maintain adequate trough levels to prevent routine bleeding (prophylaxis) will also result in only one peak level per week. Peak levels are important for hemostatic challenges such as surgery or some types of physical activity. How would one manage a situation in which a long-acting product was given a day or 2 before (with an unknown and at this time possibly unacceptable factor level) and a serious trauma occurred? Would a dose of a short-acting product be given at that point, thus resulting in “combination” therapy or would one give a dose of the patient's long-acting product? If a second dose of the long-acting factor were given, could it result in a level of factor that is potentially too high? What about the PWH who participates in athletic activities that are deemed acceptable yet pose some risk for injury? Would such a patient be better off with the shorter-acting products allowing for multiple peaks per week or would such a patient use a combination of a long-acting and short-acting product? These are but a few of the scenarios that will create new challenges in the management of hemophilia in the near future.
Finally, 2 other important issues that arise whenever new products are introduced are unforeseen adverse events and cost. Because unforeseen adverse events are by definition “unforeseen,” it is not possible to speculate on whether and what form these may take. Clearly, the clinical trials are carefully evaluating for adverse events, however, novel therapeutics have been introduced in the past that only after approval were found to have serious deleterious effects, resulting in their withdrawal from the market; examples are rofecoxib48 and fenfluramine.49 Clinicians must be highly vigilant for such possibilities and if such an event is suspected, reporting the event to the regulatory authorities (eg, the US Food and Drug Administration [FDA] and/or the European Medicines Agency [EMA]) is critical. With respect to cost, there is no doubt that novel agents with enhanced properties developed at the cost of multiple millions of dollars will exact a premium when they are priced for the market. If, for example, novel factor VIII products only prolong the half-life by 1.5- to 2-fold,3 what premium should society (health insurance) pay for this modest gain? What will patients demand and how will clinicians respond to patient demands for novel and expensive products if the benefits are only modest? These are issues that will further challenge hemophilia care providers.
New challenges in the developing world
It should be noted that the majority of PWH in the world receive no factor replacement therapy and that the mortality and morbidity in the developing world is far higher than that in countries with essentially unfettered access to factor products.50 Most of the challenges in the management of hemophilia in the developing world are not new and have been discussed previously.51 Therefore, what are the new challenges in the developing world? (Table 3 provides a list.) One major challenge facing some of the largest countries in the world with rapidly growing economies (eg, China, Brazil, and India) paradoxically is the increasing availability of factor products. The issues that this creates are multiple and include having health care professionals with the knowledge to properly and safely prescribe these products, determining how to ration the available factor among patients, and a likely increase in inhibitor patients. In less developed nations, where merely diagnosing patients is a problem, improved diagnostic capability as coagulometers become less expensive will lead to increasing recognition of hemophilia. This leads to 2 potential problems. First, patients may demand that their governments provide them with factor to treat their condition even though in many countries this will not be economically feasible. Second, because improvements in morbidity and mortality are not solely tied to factor availability, the increasing number of PWH will challenge such countries to provide services (eg, physical therapy or devices to assist ambulation) even in the absence of factor.

Another new challenge for all such nations is the development of a national database of PWH. The knowledge of who the patients are and where they live is important for several reasons. First, it allows countries to distribute both factor (if available) and, importantly, health care professionals to areas where patients live. Second, the recognition of hemophilia patients can serve to promote local advocacy by both increasing the number of PWH that are identified and that require health care services and by providing a larger voice to lobby for government to support those affected.
The above examples represent just a few of the possible new challenges in hemophilia care in the developing world. None of these should obscure the fact that most patients in the world receive no care whatsoever and many receive what could only be considered substandard care. The World Federation of Hemophilia's mission is “Treatment for All,” and those wishing to learn more about the plight of PWH are encouraged to not only visit their website at www.wfh.org, but to also get involved in the many programs that they have to achieve this mission.51
Conclusion
After more than a decade with few major changes to hemophilia care, we are entering an exciting period of innovation for the field, with novel products on the horizon that will further improve the lives of PWH. Along with the improvements in mortality and morbidity, new challenges such as obesity and osteopenia are emerging, which are likely to stimulate a period of new investigations to better understand these issues. In addition, the availability of new agents will certainly stimulate the study of novel approaches toward preventing and treating bleeding. Although the approval of these new agents may seem like the final step in a long journey, this will only be the first chapter of this new period of hemophilia care. The final chapter will be when curative therapy is available for all.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Novo Nordisk and Biogen Idec, has consulted for Novo Nordisk and Baxter, and has received honoraria from Novo Nordisk and Baxter. Off-label drug use: rFVIIa and FEIBA may include doses and indications that are outside of FDA-approved labeling (eg, the use of these agents for prophylaxis).
Correspondence
Guy Young, MD, Director, Hemostasis and Thrombosis Center, Children's Hospital Los Angeles, University of Southern California Keck School of Medicine, 4650 Sunset Blvd, Mail Stop 54, Los Angeles, CA 90027; Phone: 323-361-5507; Fax: 323-361-7128; e-mail: gyoung@chla.usc.edu.
