
Abstract
The Hodgkin and Reed/Sternberg (HRS) tumor cells of classical Hodgkin lymphoma (HL) and the lymphocyte-predominant tumor cells of nodular lymphocyte–predominant HL are both derived from germinal center B cells. HRS cells, however, have largely lost their B-cell gene-expression program and coexpress genes typical of various types of hematopoietic cells. Multiple signaling pathways show a deregulated activity in HRS cells. The genetic lesions involved in the pathogenesis of HL are only partly known, but numerous members and regulators of the NF-κB and JAK/STAT signaling pathways are affected, suggesting an important role for these pathways in HL pathogenesis. Some genetic lesions involve epigenetic regulators, and there is emerging evidence that HRS cells have undergone extensive epigenetic alterations compared with normal B cells. HRS and lymphocyte-predominant cells are usually rare in the lymphoma tissue, and interactions with other cells in the microenvironment are likely critical for HL pathophysiology. T cells represent a main population of infiltrating cells, and it appears that HRS cells both inhibit cytotoxic T cells efficiently and also receive survival signals from Th cells in direct contact with them.
Introduction
Hodgkin lymphoma (HL) is subdivided into a classical and a nodular lymphocyte–predominant form (NLPHL). Classic HL, with its subtypes of nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted HL, accounts for approximately 95% of cases, whereas NLPHL represents only 5% of cases. This distinction was made mainly based on differences in the histological picture and the morphology and immunophenotype of the tumor cells. These cells are called Hodgkin and Reed/Sternberg (HRS) cells in classical HL and lymphocyte-predominant (LP) cells in NLPHL. Gene-expression studies and analyses of the genetic lesions in the lymphoma cells further justifies that these lymphomas should be considered as separate diseases.
Cellular origin of HRS and LP cells
HRS and LP cells are both derived from germinal center (GC) B cells.1 GC B cells are antigen-activated mature B cells involved in T-cell–dependent immune responses that undergo extensive proliferation in the histological structure of the GC. A hallmark of GC B cells is the process of somatic hypermutation, through which the rearranged IgV genes are genetically modified to generate high-affinity Abs. The GC B-cell origin of LP cells is indicated by their expression of several typical GC B-cell markers (eg, BCL6, HGAL, and centerin) and by their carrying somatically mutated IgV genes with ongoing somatic hypermutation in a fraction of cases.1 Moreover, LP cells grow in follicular structures in association with follicular dendritic cells and GC Th cells, typical constituents of GC.1 HRS cells have down-regulated the expression of most B-cell–typical genes, so that immunohistochemical studies did not reveal their origin.2 However, there are now several lines of evidence proving the B-cell derivation of HRS cells (Table 1). In particular, HRS cells in nearly all cases carry clonally rearranged and somatically mutated Ig heavy- and light-chain genes.1 In approximately one-fourth of cases, clearly destructive somatic mutations (such as nonsense mutations) were detected in the rearranged IgV genes of HRS cells.1 Acquiring such mutations normally causes the immediate apoptotic death of GC B cells. Therefore, it was concluded that HRS cells derive from preapoptotic GC B cells that acquired unfavorable somatic mutations and that were rescued from apoptosis by some transforming event(s).1 Some of the transforming events in GC B cells might have happened before the HRS precursor cell entered the GC and others might occur within the GC. Moreover, this scenario does not exclude that further genetic lesions are acquired after the initial rescue of the GC B cells and the loss of the B-cell phenotype (ie, as “post-GC” B cells).
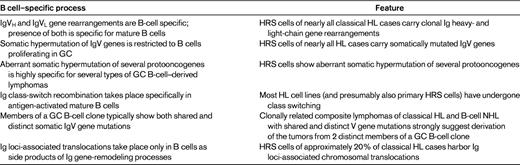
Deregulated gene expression of LP and HRS cells
HRS cells show a deregulated activity of multiple signaling pathways and transcription factors that have essential roles in the pathophysiology of these cells.1 The canonical and the noncanonical NF-κB pathway are constitutively activated in HRS cells, and NF-κB function is essential for the survival of HRS cells. The activation of NF-κB is presumably mediated in part by signaling through several cell-surface receptors, including CD40 and RANK. However, genetic lesions in HRS also play an important role (see “Somatic and germline genetic lesions”). Moreover, in approximately 40% of classical HL the HRS cells are latently infected by EBV and, in these cases, the EBV-encoded latent membrane protein 1 (LMP1) is expressed, which activates NF-κB by mimicking an activated CD40 receptor. Strong NF-κB activity has also been detected recently in LP cells of NLPHL.3
Many cytokines signal through the JAK/STAT pathway, in which upon cytokine binding, cytokine receptors activate the JAK kinases, which then phosphorylate the STAT proteins, causing translocation of STAT homo- or heterodimers into the nucleus, where they function as transcription factors. HRS cells harbor active STAT3, STAT5, and STAT6 factors, and several cytokines contribute to the activation of this pathway, including IL-13 and IL-21.1 These cytokines may be produced by other cells in the microenvironment, but some are also secreted by HRS cells, causing an autocrine stimulation.
Other signaling pathways that are typically only transiently activated in B cells upon specific activation but which are constitutively active in HRS cells include the MEK/ERK pathway and the PI3K/Akt pathway.1 HRS cells also show activation of numerous pathways and transcription factors that are not physiologically seen in normal B cells, including multiple receptor tyrosine kinases, the T-cell transcription factors Notch-1 and GATA-3, the natural killer cell factor ID2, and the myeloid receptor CSF1R.1,4,5 The activation of several of these factors contributes to the “mixed” phenotype of HRS cells, with down-regulation of most B cell–typical genes and heterogenous expression of genes of various hematopoietic cell types.
Somatic and germline genetic lesions
HRS cells of classical HL are typically aneuploid and show multiple chromosomal abnormalities.6 The cells show genomic instability, because subclonal abnormalities are usually seen in addition to clonal lesions.6 The causes for this instability are not well understood.
In addition to frequent gains and losses of parts of or whole chromosomes, chromosomal translocations have also been detected. Translocations of the MHC class II transactivator CIITA were found in 15% of HL cases.7 These translocations result in apparent nonfunctional CIITA and a consequent down-regulation of MHC class II expression,7 which may aid in an escape of HRS cells from immunosurveillance. Reciprocal translocations involving one of the Ig loci and various protooncogenes are a hallmark of many B-cell lymphomas. In classical HL, breaks in the Ig loci were detected in approximately 20% of cases.8,9 In a few cases, the partner genes have been identified as BCL1, BCL2, BCL3, BCL6, REL, and MYC. However, for most cases, it is still unclear which genes are involved in the translocations with the Ig loci. In any case, the pathogenetic role of these translocations is mostly unclear. Translocations involving the Ig loci usually function through the strong activity of the Ig enhancer elements, which cause a deregulated and constitutive expression of the protooncogenes translocated into the Ig loci. However, as HRS cells have largely down-regulated the expression of their Ig genes, one would not expect an Ig enhancer-driven deregulation of the genes translocated into the Ig loci in HRS cells. Perhaps these translocations were important in the precursor cells of the HRS cells that still had a B-cell phenotype and hence active Ig loci, but additional transforming events later in HL pathogenesis replaced the oncogenic role of these genes.
The search for somatic mutations within oncogenes and tumor-suppressor genes in HRS cells is much hampered by the rarity of the HRS cells in the lymphoma tissue. Candidate genes were initially studied in the few HL cell lines available and if mutations were found, microdissected primary HRS cells were then analyzed. Based on the detection of a strong and constitutive NF-κB activity with prosurvival functions in HRS cells, members of this pathway were studied for mutations. Multiple genetic lesions were indeed found in members of this pathway (Table 2). The gene NFKBIA, encoding IκBα, the main inhibitor of the canonical NF-κB pathway, is inactivated by mutations in approximately 10%-15% of classical HL cases.1,10 Mutations were also found in NFKBIE, encoding the NF-κB inhibitory factor IκBϵ.1 A20, which is encoded by the TNFAIP3 gene and also functions as an inhibitor of NF-κB, is affected by inactivating mutations in approximately 40% of classical HL cases.11,12 Interestingly, TNFAIP3 mutations are mostly mutually exclusive with the presence of EBV in the HRS cells, suggesting that these are alternative pathogenetic mechanisms contributing to the strong NF-κB activity in HRS cells.12 Somatic mutations were also identified in 2 other negative regulators of NF-κB signaling, CYLD and TRAF3, but these seem to be relatively rare (Table 2).13,14 The gene of the NF-κB factor REL is affected by genomic gains and amplifications in approximately 50% of HL cases, and these gains are associated with an increased level of the REL protein.1 MAP3K14 (NIK), encoding a main factor of the alternative NF-κB pathway, also frequently shows gains in HRS cells and may thus function as an oncogene in HRS cells.13,15 Because most studies of the NF-κB factors and regulators were performed with separate collections of cases of classical HL, we know little about the cooccurrence of these lesions in primary HRS cells. However, the study of HL cell lines revealed that many of the lines carry lesions in 2 or even 3 of the genes. Therefore, it appears that they often coexist and that lesions in more than one factor and of both NF-κB pathways are needed for the strong NF-κB activity and the pathogenesis of HRS cells.
Somatic genetic lesions in HRS and LP cells
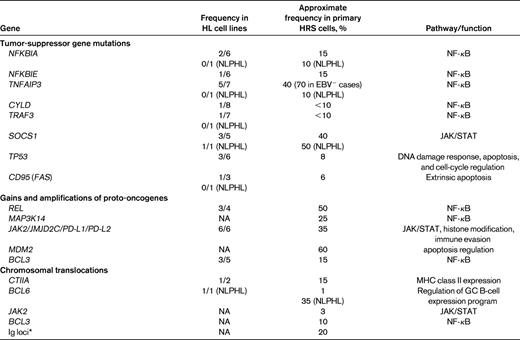
Only genes in that mutations were found are listed. Numbers refer to HRS cells if not otherwise specified. Note that “<10” refers to no mutations found in 8 of 10 cases analyzed, and the 10% for NFKBIA and TNFAIP3 in NLPHL for 1 case each among 10 cases tested with a mutation.
NA refers to not analyzed.
*With various partners (ie, BCL1, BCL2, BCL3, BCL6, and MYC), mostly still unknown.
The JAK/STAT pathway is another signaling pathway for which several genetic lesions have been identified in HRS cells. SOCS1, encoding the main inhibitor of JAK/STAT signaling, is inactivated by mutations in approximately 40% of classical HL cases. The chromosomal region 9p24, which includes JAK2, frequently shows genomic gains and, in rare instances, JAK2 is involved in chromosomal translocations (Table 2). Interestingly, the amplified region in 9p24 usually also includes the genes programmed death 1 (PD1) ligand 1 and 2 (PD-L1 and PD-L2) and the histone demethylase gene JMJD2C.16,17 Because PD-L1 and PD-L2 may contribute to escape of the HRS cells from immunosurveillance and because inhibition of JMJD2C is toxic for HRS cells,17 a single genetic lesion, the gains of 9p24, involves at least 4 pathogenetically relevant genes.
Several other genetic lesions detected in HRS cells affect regulators of apoptosis, including MDM2, TP53, and CD95 (Table 2).
Only a few genetic lesions are known for LP cells of NLPHL (Table 2). LP cells in approximately 35% of NLPHL cases carry chromosomal translocations of the protooncogene BCL6, involving various translocation partners, including the Ig loci.1 SOCS1 is affected by mutations in approximately 40% of cases. Although LP cells show a similar or even higher NF-κB activity to HRS cells,3 the mechanisms for this deregulation appear to be quite distinct in these cells. No inactivating mutations were found in NFKBIA and TNFAIP3 in LP cells,18 and REL gains have also not been identified. In addition, LP cells are only EBV infected in rare cases, so a contribution of LMP1 to NF-κB activation, as seen in EBV+ HRS cells, also does not play a role in LP cells.
HL is one of the lymphomas with the strongest familial association. It is therefore conceivable that germline polymorphisms or mutations may play a role in a fraction of cases. Indeed, in a family with 4 members affected by NLPHL, a truncating germline mutation in the NPAT gene was found in all patients.19 Moreover, in sporadic cases of HL, a germline deletion of one amino acid of NPAT was found at increased frequency in HL patients compared with controls (odds ratio, 4.11; P = .018).19 The function of NPAT is not well understood, but it appears to be involved in regulation of the cell cycle and the activity of the ATM promoter. The gene KLHDC8B was found to be affected by a constitutional translocation in another family, with several members developing HL.20 A germline polymorphism in the 5′ untranslated region of KLHDC8B causing reduced translation was identified in other families with multiple HL patients.20 Remarkably, down-regulation of KLHDC8B in HeLa cells caused an increased frequency of binucleated cells, so that these alterations might play a role in the generation of bi- or multinucleated Reed/Sternberg cells.20 In a genome-wide association study of HL patients, risk loci were detected at 2p16.2, 8q24.21, and 10p14. The odds ratios for these associations are relatively low, but it is remarkable that these loci include the genes REL, PVT1, and GATA3, respectively.21 REL was already discussed as a member of the NF-κB family with frequent somatic gains in HRS cells, PVT1 is involved in translocations in lymphoid tumors, and GATA3 is a T-cell transcription factor with aberrant expression and activity in HRS cells.5
Epigenetic alterations in HRS cells
Deregulated gene expression in tumor cells is caused not only by genetic lesions, but often also by epigenetic alterations. Most studies in HL have focused on the methylation of CpG sites in the promoter region of genes, which typically mediates silencing of gene transcription. Several known tumor-suppressor genes were studied for silencing via DNA methylation in HL cell lines and/or primary HRS cells. Frequent methylation was found for the tumor-suppressor genes CDKN2C (p18-INK4c), CHEK2, and RASSF1A.22–24 In addition, the gene of the proapoptotic cell-adhesion molecule IGSF4 is frequently hypermethylated in HRS cells, and its ectopic reexpression in HL cell lines decreased cell survival, supporting a pathogenetic role of IGSF4 silencing in HRS cells.25 KLF4, a transcription factor implicated in the regulation of B-cell maturation and the suppression of B-cell proliferation, is down-regulated in HRS cells and this is associated with a hypermethylated status of the gene.26 KLF4 reexpression in HL cell lines was shown to cause massive cell death, supporting its tumor-suppressor function.26
Epigenetic alterations are also involved in the down-regulation of B-cell–typical genes in HRS cells. For example, CD79B, BOB1, and SYK were found to be silenced by DNA methylation in HRS cells.27,28 These findings were recently extended by a global analysis of DNA methylation in HL cell lines, which revealed that many genes specifically methylated in HL cell lines compared with normal B cells and other GC B-cell–derived lymphomas represented components of B-cell functions.29 The epigenetic silencing of B-cell genes in HRS cells also involves histone modifications, because HL cell lines show hypoacetylation at histone H3 and H3K27 trimethylation of many B-cell genes.30 These histone marks are typical for transcriptionally inactive chromatin. There are contradictory reports as to whether global demethylation of DNA in HL cell lines restores a B-cell phenotype in these cells.28,31 Interestingly, histone acetylation and DNA demethylation of lymphoma cell lines with a typical B-cell phenotype induced down-regulation of the B-cell phenotype, up-regulation of non–B-cell genes, and acquisition of gene-expression features of HRS cells.31 This indicates that the up-regulation of non–B-cell genes in the generation of HRS cells is partly mediated by epigenetic mechanisms.
Considering the multiple epigenetic changes in HRS cells, it is an intriguing issue to find out whether (and how) these alterations are associated with altered expression and activity of regulators of DNA methylation and/or histone modifications. Indeed, deregulated expression of several of such regulators is known for HRS cells. Polycomb group (PcG) proteins are important chromatin modifiers that mainly function by establishing repressing chromatin marks, for example, to silence genes of differentiated cells in stem and precursor cells. HRS cells show an up-regulated expression of members of both PcG-repressive complexes, including the PcG1 members BMI1 and RING1, and the PcG2 complex members EZH2, EED, and YY2.32,33 PcG1 and PcG2 complex expression is also seen in normal GC B cells, but in these cells, the 2 complexes show a mutually exclusive expression in the 2 main subsets of GC B cells, the proliferating centroblasts (PcG2 expression) and the nonproliferating centrocytes (PcG1 expression), whereas they are coexpressed in HRS cells.33 To what extent the PcG factors are involved in epigenetic alterations in HRS cells is not clear. One study reported that a large fraction of genes with de novo methylation in HRS cells are enriched for PcG2 complex targets,34 whereas another study showed only little enrichment of PcG targets among genes specifically hypermethylated in HL cell lines.29
Adding further complexity to the picture of deregulated epigenetic regulators in HRS cells, it was reported recently that a counter player of PcG2 complex activity, KDM6B (previously also called JMJD3) is aberrantly up-regulated in HRS cells.35 This factor is a H3K27me3 demethylase, so it can remove the repressive methylation marks from histone H3K27. KDM6B appears to be active in HRS cells, because its silencing in HL cell lines increased the H3K27 methylation of known KDM6B target genes.35
Striking evidence for the pathogenetic role of epigenetic alteration in HRS cells was reported recently by Lamprecht et al,4 who showed that the myeloid-specific factor and protooncogene colony stimulating factor 1 receptor (CSF1R) is aberrantly expressed in HRS cells due to derepression of an endogenous long terminal repeat located upstream of the CSF1R promoter. This derepression is mediated by epigenetic mechanisms and linked to down-regulation of the chromatin modifier CBFA2T3.4
The causes for the altered expression of epigenetic regulators in HRS cells are mostly unclear. However, in at least some cases, genetic lesions are the cause. For example, the frequent 9p24 amplification in HRS cells involves not only the JAK2 gene, but also the gene JMJD2C, encoding a histone demethylase.17 Strikingly, JAK2 functions not only as an activator of STAT transcription factors, but also as an epigenetic regulator through its kinase activity on histones. Therefore, the 9p24 amplification deregulates 2 histone modifiers, and the pathogenetic relevance of this deregulation is evident from the observation that silencing of these factors is toxic for HL cell lines.17
Interaction of HRS cells with T cells
Among the various cell types that are found in the HL microenvironment, T cells are of particular interest because they usually represent the largest population of infiltrating cells and are the predominant cells in direct contact with the HRS and LP cells. In NLPHL, the T cells rosetting around the LP cells show phenotypic markers of follicular Th cells, including expression of CD57, PD1, and BCL6.36 This fits with the histological appearance of NLPHL, in which LP cells are embedded in a GC-like environment.
In classical HL, HRS cells are also surrounded directly by CD4+ T cells. However, these cells do not have a GC Th-cell phenotype and they seem to be a mixed population of Th cells and regulatory T (Treg) cells.37–39 The Th cells have largely a Th2 phenotype, which is characteristic of T cells supporting B cells in humoral immune responses. HRS cells in approximately 40% of cases down-regulated MHC class II expression,7,40 which is essential for cognate interactions between T cells and B cells. Therefore, it is not well understood how these T cells interact with the HRS cells. Nevertheless, T-cell rosetting around HRS cells is seen in most cases of classical HL, and HRS cells attract chemokine receptor CCR4+ Th and Treg cells actively by the secretion of large amounts of the corresponding chemokine CCL17.37,41 In addition, HL cell lines in culture spontaneously form clusters with CD4+ T cells. These features indicate that the HRS-cell/T-cell interaction is important for HRS cells, and this is further supported by the observation that reestablishment of the CD4+ T-cell pool in AIDS patients by antiretroviral therapy is associated with an increased incidence of HL.42 Potential factors for the interaction of CD4+ cells with HRS cells include production of the HRS cell growth factor IL-13 by the T cells,36 stimulation of HRS cells through interaction between CD28 expressed by the T cells and CD80 and CD86 expressed by HRS cells, and interaction between CD40 on HRS cells and CD40L on rosetting T cells (Figure 1).

Interactions between HRS cells and tumor-infiltrating T cells in the microenvironment of classical HL. Shown are surface molecules and secreted factors through which HRS cells and HL-infiltrating T cells interact. Further details are given in the text.
Interactions between HRS cells and tumor-infiltrating T cells in the microenvironment of classical HL. Shown are surface molecules and secreted factors through which HRS cells and HL-infiltrating T cells interact. Further details are given in the text.
Tumor-infiltrating Treg cells appear to play an important role in the rescue of HRS cells from an attack by cytotoxic T and natural killer cells (Figure 1). Treg cells produce the immunosuppressive cytokine IL-10, and they express the T-cell–inhibitory receptor cytotoxic T-lymphocyte associated protein 4 (CTLA4).38 It is likely that additional, yet-to-be defined factors further contribute to the suppressive activity of the Treg cells in the HL microenvironment. The function of Tregs in inhibiting cytotoxic T cells has been demonstrated in in vitro coculture studies.38 HRS cells appear not only to attract Treg cells into the lymphoma microenvironment, but to also be capable of directly influencing the differentiation of CD4+ T cells into Treg cells.43
The inhibition of cytotoxic T cells and natural killer cells in classical HL is not only mediated through Treg cells, but also directly by the HRS cells. HRS cells produce the immunosuppressive factors IL-10, TGFβ, galectin-1, tissue inhibitor of metalloproteinase 1 (TIMP1), and prostaglandin E2 (PGE2).44–47 They also express the PD1 ligands 1 and 2, which inhibit PD1+ T cells, and express CD95 ligand, which can induce apoptosis of CD95+-activated CD8+ and Th1 T cells (Figure 1).44,48
Acknowledgments
The author thanks Martin-Leo Hansmann and all members of the group for many stimulating discussions and apologizes to all colleagues whose work could not be cited adequately due to space and reference restrictions.
The author's work discussed herein was supported by the Deutsche Forschungsgemeinschaft, the Deutsche Krebshilfe, and the Wilhelm-Sander Foundation.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Ralf Küppers, Institute of Cell Biology (Cancer Research), University of Duisburg-Essen, Medical School, Virchowstr 173, 45122 Essen, Germany; Phone: 0049-201-7233384; Fax: 0049-201-7233386; e-mail: ralf.kueppers@uk-essen.de.
