
Abstract
Because of the central role of platelets in cardiovascular atherothrombosis, there is a well-established therapeutic role for antiplatelet therapy that includes aspirin (a cyclooxygenase 1 [COX1] inhibitor), clopidogrel (an antagonist of the ADP P2Y12 receptor), and the GPIIb-GPIIIa (αIIbβ3) antagonists. However, there remains a significant incidence of arterial thrombosis in patients treated with currently available antiplatelet therapy. Novel P2Y12 antagonists such as the recently US Food and Drug Administration (FDA)–approved prasugrel, along with ticagrelor, cangrelor, and elinogrel, have advantages over clopidogrel, including more rapid, less variable, and more complete inhibition of platelet function. Currently ongoing phase 3 studies will determine whether these new P2Y12 antagonists will result in better and/or more rapid antithrombotic effects than clopidogrel, without an unacceptable increase in hemorrhagic or other side effects, as has been recently reported in some clinical settings for prasugrel and ticagrelor. Antagonists of the thrombin receptor protease-activated receptor 1 (PAR1) are also undergoing phase 3 trials, and many other novel antiplatelet agents are under investigation as antithrombotic agents.
Introduction
In normal hemostasis, platelets prevent hemorrhage after injury and thereby preserve vascular integrity. However, vascular damage such as rupture of an atherosclerotic plaque can result in a platelet-dependent thrombus that may lead to vascular occlusion with resultant hypoxia and infarction of distal tissues. Therefore, thrombotic occlusion of a coronary artery results in acute myocardial infarction and thrombotic occlusion of a cerebral artery results in acute ischemic stroke. Because of this central role of platelets in arterial thrombosis, antiplatelet therapy has proven to be beneficial in this setting.1
Therapies targeting key pathways of platelet activation (Figure 1), including thromboxane A2 synthesis, ADP-mediated signaling, and integrin αIIbβ3 (GPIIb-GPIIIa), have an established role in the treatment of cardiovascular arterial disease. However, there are limitations to these agents (including aspirin, clopidogrel, and GPIIb-GPIIIa antagonists) and there remains a significant incidence of arterial thrombosis in patients treated with currently available antiplatelet therapy. Limitations of current therapies include weak inhibition of platelet function (eg, aspirin), blockade of only one pathway of ADP-mediated signaling (eg, clopidogrel), slow onset of action (eg, clopidogrel), interpatient response variability with poor inhibition of platelet function in some patients (eg, clopidogrel), inability to transform the success of intravenous GPIIb-GPIIIa antagonist therapy into successful oral GPIIb-GPIIIa antagonist therapy, and the inability to completely separate a reduction in thrombotic events from an increase in bleeding events.

Platelet function and molecular targets of antiplatelet agents. Initial platelet adhesion to damaged vessel walls is mediated by exposed collagen binding to platelet surface GPVI and integrin α2β1 and by VWF binding to the platelet surface GPIb-X-V complex. Thrombin, generated by the coagulation cascade, is a potent activator of human platelets via 2 platelet surface receptors: PAR1 and PAR4. Three groups of platelet surface receptors provide important positive feedback loops for platelet activation: (1) P2Y1 and P2Y12 receptors are stimulated by ADP released from platelet dense granules; (2) 5-HT2A receptors are stimulated by serotonin (5-HT) released from platelet-dense granules; (3) the thromboxane prostanoid (TP) receptor is stimulated by TXA2 generated by the platelet COX1-dependent signaling pathway. Platelet-to-platelet aggregation is mediated by fibrinogen and, at high shear flow, VWF binding to the activated molecular conformation of GPIIb-GPIIIa. Platelet-monocyte adhesion is initially mediated by the binding of platelet surface P-selectin (which is only expressed on the platelet surface after platelet degranulation) to its constitutively expressed counterreceptor PSGL-1 on the monocyte surface. Molecular targets of FDA-approved antiplatelet agents are shown in blue. Unfractionated heparin, low-molecular-weight heparin, and direct thrombin inhibitors such as lepirudin, argatroban, bivalirudin, and dabigatran, unlike PAR1 antagonists, are anticoagulants rather than specific antiplatelet drugs. However, their inhibition of thrombin results in less platelet activation. Molecular targets of antiplatelet agents in clinical development are shown in green. Investigational strategies for novel antiplatelet agents are shown in red. LMWH, low molecular weight heparin; PG, prostaglandin; PSGL-1; P-selectin glycoprotein ligand 1; TX, thromboxane; UFH, unfractionated heparin. (Reproduced with permission from Michelson.1 )
Platelet function and molecular targets of antiplatelet agents. Initial platelet adhesion to damaged vessel walls is mediated by exposed collagen binding to platelet surface GPVI and integrin α2β1 and by VWF binding to the platelet surface GPIb-X-V complex. Thrombin, generated by the coagulation cascade, is a potent activator of human platelets via 2 platelet surface receptors: PAR1 and PAR4. Three groups of platelet surface receptors provide important positive feedback loops for platelet activation: (1) P2Y1 and P2Y12 receptors are stimulated by ADP released from platelet dense granules; (2) 5-HT2A receptors are stimulated by serotonin (5-HT) released from platelet-dense granules; (3) the thromboxane prostanoid (TP) receptor is stimulated by TXA2 generated by the platelet COX1-dependent signaling pathway. Platelet-to-platelet aggregation is mediated by fibrinogen and, at high shear flow, VWF binding to the activated molecular conformation of GPIIb-GPIIIa. Platelet-monocyte adhesion is initially mediated by the binding of platelet surface P-selectin (which is only expressed on the platelet surface after platelet degranulation) to its constitutively expressed counterreceptor PSGL-1 on the monocyte surface. Molecular targets of FDA-approved antiplatelet agents are shown in blue. Unfractionated heparin, low-molecular-weight heparin, and direct thrombin inhibitors such as lepirudin, argatroban, bivalirudin, and dabigatran, unlike PAR1 antagonists, are anticoagulants rather than specific antiplatelet drugs. However, their inhibition of thrombin results in less platelet activation. Molecular targets of antiplatelet agents in clinical development are shown in green. Investigational strategies for novel antiplatelet agents are shown in red. LMWH, low molecular weight heparin; PG, prostaglandin; PSGL-1; P-selectin glycoprotein ligand 1; TX, thromboxane; UFH, unfractionated heparin. (Reproduced with permission from Michelson.1 )
Aspirin (Figure 1 and Table 1) remains the cornerstone of antiplatelet therapy because of its proven benefit and very low cost.1 GPIIb-GPIIIa antagonists (Figure 1 and Table 1), even in the current era of elective percutaneous coronary intervention (PCI) performed with stents and thienopyridines, still provide clinical benefits (reduced nonfatal acute myocardial infarction without a notable increase in major bleeding but an increase in minor bleeding and no reduction in all-cause mortality).2 However, aspirin and GPIIb-GPIIIa antagonists will not be specifically discussed in this article. Rather, the focus will be on recent advances in antiplatelet therapy.

ADP receptor antagonists
ADP, an important platelet agonist in vivo, has 2 types of receptors in the platelet plasma membrane: P2Y1 and P2Y12 (Figure 1).3 P2Y1 is a 7-transmembrane G-protein–coupled receptor linked to Gq. The end result of ADP signaling through its P2Y1 receptor is calcium mobilization, platelet shape change, and rapidly reversible platelet aggregation. P2Y12 is also a 7-transmembrane domain receptor, but it is linked to a Gi protein and lowering of cyclic adenosine monophosphate. The end result of ADP signaling through its P2Y12 receptor is amplification of stable platelet aggregation and secretion.3
Clopidogrel
Clopidogrel (Plavix; sanofi aventis/Bristol-Myers Squibb) is a thienopyridine (Table 1) that is metabolized through cytochrome P450 in the liver. Its active metabolite irreversibly antagonizes the P2Y12 receptor (Figure 1).3 Clopidogrel is given orally as a daily dose. The clinical benefit of adding clopidogrel to aspirin therapy has been demonstrated by large, multicenter, randomized controlled trials such as CURE (Clopidogrel in Unstable Angina to Prevent Recurrent Events) in patients with acute coronary syndrome (ACS), unstable angina, or non-ST elevation myocardial infarction; PCI-CURE (Percutaneous Coronary Intervention CURE) and CREDO (Clopidogrel for the Reduction of Events During Observation) in patients undergoing PCI; and COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) and CLARITY-TIMI 28 (Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myocardial Infarction 28) in patients with ST-elevation myocardial infarction.3 However, in patients with stable cardiovascular disease or in asymptomatic patients with multiple cardiovascular risk factors, the CHARISMA (Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance) trial4 demonstrated that the combination of clopidogrel plus aspirin was not significantly more effective than aspirin alone in reducing the rate of myocardial infarction, stroke, or death from cardiovascular causes. Furthermore, the risk of moderate-to-severe bleeding was increased.4 In a retrospective analysis of the CHARISMA trial, dual antiplatelet therapy with clopidogrel and aspirin in the primary prevention subgroup of patients was associated with an increase in cardiovascular death.5 The cause of this apparent harm has not been elucidated.
Monitoring of clopidogrel by platelet function assays reveals interpatient response variability.6 Furthermore, there is evidence that a poor response to clopidogrel in such in vitro assays (referred to as “clopidogrel resistance,” “clopidogrel hyporesponsiveness,” or, preferably, “high on-treatment platelet reactivity to ADP”7 ) is associated with major adverse cardiovascular events.7 This observation, together with the relatively slow onset of action of clopidogrel,3 has led to the development of novel P2Y12 antagonists and other antiplatelet agents that are either US Food and Drug Administration (FDA)–approved (prasugrel) or in clinical development (Table 2).
Carriers of the common reduced-function CYP2C19 allele have a lower level of the active metabolite of clopidogrel, diminished platelet inhibition, and a higher rate of major adverse cardiovascular events than noncarriers.8,9 In contrast, prasugrel, because of its different metabolism (via esterases and less dependence upon CYP enzymes),10 is unaffected by the reduced-function CYP2C19 allele,11 and ticagrelor12 and cangrelor are unaffected because they do not require metabolism. However, among patients with ACS or atrial fibrillation, the effect of clopidogrel compared with placebo is consistent, irrespective of CYP2C19 loss-of-function carrier status.13 The explanation for this finding may be that previous studies that have shown an attenuated benefit of clopidogrel among carriers of loss-of-function alleles did not include a randomized control group, and therefore potential pleiotropic effects of loss-of-function alleles (ie, independent of their effects on active metabolite levels of clopidogrel) could not be excluded.13
Pharmacokinetic and pharmacodynamic studies, using platelet assays as surrogate end points, have suggested that the concomitant use of clopidogrel and a proton pump inhibitor (both of which are metabolized through cytochrome P450) reduces the antiplatelet effects of clopidogrel.14 The strongest evidence for an interaction is between omeprazole and clopidogrel. However, it has not been established that changes in these surrogate end points translate into clinically meaningful differences.14
Prasugrel
Prasugrel (Effient; Eli Lilly/Daiichi Sankyo) is an orally administered thienopyridine prodrug that, like clopidogrel, is metabolized via liver cytochrome P450 (Table 2)3 to its active metabolite, which irreversibly inhibits the platelet P2Y12 receptor (Figure 1). However, metabolism of prasugrel to its active metabolite is much more efficient than that of clopidogrel, because it is metabolized via esterases and is less dependent upon CYP enzymes.10 As a result, a prasugrel 60-mg loading dose results in a much more rapid, potent, and consistent inhibition of platelet function than the standard clopidogrel loading dose of 300 mg and the increasingly used clopidogrel loading dose of 600 mg.15 Furthermore, a maintenance dose of prasugrel of 10 mg daily results in a more potent and consistent inhibition of platelet function than both the standard clopidogrel maintenance dose of 75 mg daily and clopidogrel 150 mg daily.15
TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel—Thrombolysis in Myocardial Infarction 38), a 13 608-patient phase 3 trial, demonstrated that in patients with ACS with scheduled PCI, prasugrel (60-mg loading dose and a 10-mg daily maintenance dose), compared with approved doses of clopidogrel (300-mg loading dose and a 75-mg daily maintenance dose), significantly reduced rates of ischemic events, including stent thrombosis, but with an increased risk of major bleeding, including fatal bleeding.16 The primary efficacy end point occurred in 12.1% of patients receiving clopidogrel and in 9.9% of patients receiving prasugrel. In the prasugrel group, there were also significant reductions in the rates of myocardial infarction, urgent target-vessel revascularization, and stent thrombosis. Major bleeding was observed in 2.4% of patients receiving prasugrel and in 1.8% of patients receiving clopidogrel. Also greater in the prasugrel group was the rate of life-threatening bleeding, including nonfatal bleeding and fatal bleeding. A post hoc subgroup exploratory analysis of the data identified 3 subgroups of interest who had less clinical efficacy and greater absolute levels of bleeding than the overall cohort, resulting in less net clinical benefit (age > 75 years, body weight < 60 kg) or in clinical harm (history of stroke or transient ischemic attack).16
The TRITON-TIMI 38 platelet substudy showed that prasugrel results in greater inhibition of ADP-mediated platelet function than clopidogrel in patients with ACS, supporting the hypothesis that greater platelet inhibition leads to a lower incidence of ischemic events and more bleeding both early and late after PCI.17 In a prespecified TRITON-TIMI 38 study of 3534 STEMI patients undergoing PCI, prasugrel was more effective than clopidogrel for the prevention of ischemic events without an apparent excess in bleeding.18 In TRITON-TIMI 38, diabetic subjects had a greater prasugrel-induced reduction in ischemic events than nondiabetic subjects without an observed increase in TIMI major bleeding.19
Largely based on the TRITON-TIMI 38 trial, prasugrel is now approved in the United States and Europe for the prevention of atherothrombotic events in patients with ACS undergoing PCI. Because of the bleeding risk, the prescribing information contains a “black box warning” to not use prasugrel in patients with a history of transient ischemic attack or stroke or in patients ≥ 75 years of age. Body weight < 60 kg is also a risk factor for bleeding. An additional phase III clinical trial (TRILOGY ACS, www.clinicaltrials.gov/ct2/show/NCT00699998) of prasugrel is in progress in the setting of medically managed ACS rather than the PCI in ACS setting of TRITON-TIMI 38. In TRILOGY ACS, the prasugrel dose is 5 or 10 mg daily depending on the patient's age and weight.
Ticagrelor
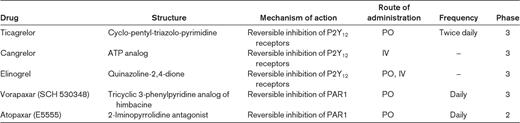
Ticagrelor (AZD6140; AstraZeneca) is an investigational P2Y12 antagonist (Figure 1 and Table 2) that is administered orally twice a day. In contrast to ticlopidine, clopidogrel, and prasugrel (all of which are thienopyridine prodrugs), ticagrelor is a cyclo-pentyl-triazolo-pyrimidine and is a direct and reversible P2Y12 antagonist.3 Like prasugrel, ticagrelor acts more rapidly and is a more potent inhibitor of platelets than clopidogrel.3,20 Ticagrelor therapy overcomes nonresponsiveness to clopidogrel, and its antiplatelet effect is the same in responders and nonresponders.21 After drug discontinuation, the offset of platelet inhibition is faster for ticagrelor than for clopidogrel.22 The occurrence of dyspnea and ventricular pauses is greater, in an apparently dose-dependent manner, with ticagrelor compared with clopidogrel. However, the dyspnea does not appear to be associated with any adverse change in cardiac or pulmonary function,23 and the ventricular pauses (which were shown to be predominantly asymptomatic and sinoatrial node in origin) are not associated with apparent clinical consequences.24
PLATO (Platelet Inhibition and Patient Outcomes) was a multicenter, double-blind, randomized trial comparing ticagrelor (180-mg loading dose and 90 mg twice daily thereafter) and clopidogrel (300- to 600-mg loading dose and 75 mg daily thereafter) for the prevention of cardiovascular events in 18 624 patients admitted to the hospital with an ACS, with or without ST-segment elevation.25 At 12 months, the primary end point—a composite of death from vascular causes, myocardial infarction, or stroke—had occurred in 9.8% of patients receiving ticagrelor compared with 11.7% of those receiving clopidogrel (hazard ratio, 0.84; 95% confidence interval [95%CI], 0.77-0.92; P < .001). Predefined hierarchical testing of secondary end points showed significant differences in the rates of other composite end points, as well as myocardial infarction alone (5.8% in the ticagrelor group vs 6.9% in the clopidogrel group, P = .005) and death from vascular causes (4.0% vs 5.1%, P = .001) but not stroke alone (1.5% vs 1.3%, P = .22). The rate of death from any cause was also reduced with ticagrelor (4.5% vs 5.9% with clopidogrel; P < .001). No significant difference in the rates of major bleeding was found between the ticagrelor and clopidogrel groups (11.6% and 11.2%, respectively, P = .43), but ticagrelor was associated with a higher rate of major bleeding not related to coronary-artery bypass grafting (4.5% vs 3.8%, P = .03), including more instances of fatal intracranial bleeding and fewer of fatal bleeding of other types.25
Although the FDA Cardiovascular and Renal Drugs Advisory Committee recommended approval of ticagrelor for clinical use in July 2010, ticagrelor at the time of writing is still not FDA approved. The main concern is the “US paradox”: in the 18 758 subjects enrolled in PLATO worldwide, ticagrelor showed overall benefit compared with clopidogrel, but in the 1814 subjects enrolled in the United States and Canada, ticagelor showed no benefit compared with clopidogrel, with a trend in favor of clopidogrel.26 It remains uncertain whether the explanation for the US paradox is chance, the result of a higher aspirin dose in US and Canadian patients, and/or another factor(s).26
Cangrelor
Cangrelor (The Medicines Company) is an investigational, direct-acting, reversible P2Y12 antagonist (Figure 1 and Table 2).3 Unlike each of the P2Y12 antagonists described above, cangrelor is administered intravenously with a rapid reversal of cangrelor's effects after the end of the infusion. Like prasugrel and ticagrelor, cangrelor results in a more rapid onset of action and greater degree of platelet inhibition than clopidogrel, and showed no significant increase in major bleeding compared with clopidogrel in phase 2 studies.3 Cangrelor subsequently underwent 2 phase 3 trials, CHAMPION-PCI and CHAMPION-PLATFORM, which were stopped early for lack of efficacy.27,28 The CHAMPION-PHOENIX phase 3 trial in patients undergoing PCI is still enrolling (www.clinicaltrials.gov/ct2/show/NCT01156571). In addition, cangrelor is being studied as a bridge for patients on clopidogrel who need to terminate treatment before surgery (www.clinicaltrials.gov/ct2/show/NCT00767507).
Elinogrel
Elinogrel (PRT060128; Novartis) is an investigational, direct-acting, reversible P2Y12 antagonist with a novel structure (Figure 1 and Table 2).3 Elinogrel can be administered orally or intravenously. High platelet reactivity to ADP in patients on clopidogrel can be reversibly overcome by elinogrel.29 After successful phase 2 studies (eg, INNOVATE-PCI, presented in abstract form at the European Society of Cardiology 2010 Congress), a phase 3 trial of elinogrel is currently in the planning stage.
PDE inhibitors
Dipyridamole
Dipyridamole is a pyrimidopyrimidine derivative with both antiplatelet and vasodilator properties (Table 1).1 The antiplatelet effects of dipyridamole have been reported to be due to several mechanisms of action, including inhibition of cyclic nucleotide phosphodiesterase (PDE) and blockade of adenosine uptake, both of which result in increased intraplatelet cyclic adenosine monophosphate, thereby inhibiting signal transduction (Figure 1). A modified-release formulation of 200-mg dipyridamole with improved bioavailability has been developed in association with low-dose (25 mg) aspirin (Aggrenox; Boehringer Ingelheim). Aggrenox is FDA approved for stroke prevention based on the results of the European Stroke Prevention Study 2 (ESPS-2)30 and the European Stroke Prevention Reversible Ischemia Trial (ESPRIT).31 However, Aggrenox was not superior to clopidogrel in the treatment of recurrent stroke in the Prevention Regimen for Effectively Avoiding Second Strokes (PRoFESS) trial.32
Cilostazol
Cilostazol (Pletal; Otsuka) is an oral selective cyclic nucleotide PDE3 inhibitor with antiplatelet, vasodilatory, and antimitogenic effects (Figure 1 and Table 1).1 Cilostazol is FDA approved for intermittent claudication, and has been investigated for use in PCI and stroke. Like aspirin and clopidogrel, cilostazol appears to be effective and safe in reducing the risk of restenosis and repeat revascularization after PCI, but available evidence is limited by small study effects.33 Despite the greater reduction of platelet reactivity by the addition of cilostazol to conventional dual antiplatelet therapy with aspirin and clopidogrel, triple antiplatelet therapy did not show superiority in reducing a composite end point of adverse cardiovascular outcomes after drug-eluting stent implantation.34 Compared with aspirin, cilostazol inhibited progression of carotid intima media thickness, an established surrogate marker of cardiovascular events in patients with type 2 diabetes mellitus.35 Unfortunately, cilostazol side effects (including headaches, gastrointestinal symptoms, and skin rash) lead to discontinuation of the drug by ∼ 15% of patients.36
PAR1 antagonists
Thrombin, generated by the coagulation cascade, is a potent activator of human platelets via actions on 2 platelet surface G-protein coupled receptors, protease-activated receptor 1 (PAR1) and PAR4 (Figure 1).1 PAR1 antagonists are therefore inhibitors of thrombin-induced platelet activation but not thrombin-induced cleavage of fibrinogen, the final step in coagulation.
Vorapaxar
Vorapaxar (SCH 530348; Schering-Plough), a synthetic tricyclic 3-phenylpyridine analog of himbacine, is an orally administered, rapidly absorbed, high-affinity reversible PAR1 antagonist (Figure 1 and Table 2). A phase 2 trial demonstrated that vorapaxar inhibited PAR1 thrombin receptor activating peptide (TRAP)–induced platelet aggregation in a dose-dependent manner, was generally well tolerated, and did not cause an increase in major bleeding, even when administered concomitantly with aspirin and clopidogrel.37 Two phase 3 trials of vorapaxar are currently in progress: TRA-CER (www.clinicaltrials.gov/ct2/show/NCT00527943) and TRA 2P-TIMI 50 (www.clinicaltrials.gov/ct2/show/NCT00526474). The Data and Safety Monitoring Committee recently recommended stopping 1 of the 3 arms of TRA 2P-TIMI 50 (because of an excess in intracranial hemorrhage in patients with a history of ischemic stroke randomized to vorapaxar) and closing out the TRACER study (because a sufficient number of events were collected to test the primary hypothesis of the study).38
Atopaxar
The overall results of large phase 2 trials of another PAR1 antagonist, atopaxar (E5555, Eisai; Figure 1 and Table 2), could be considered sufficiently positive to embark on a phase 3 trial.38–40 However, the numerically higher incidence of major bleeding complications, liver dysfunction, and QTc prolongation and the lack of a convincing dose-related trend for bleeding risk and efficacy may be reasons for caution.38–40
Other Investigational Approaches

Conclusions
Because of the central role of platelets in cardiovascular atherothrombosis, there is a well-established therapeutic role for antiplatelet therapy, including the cyclooxygenase 1 (COX1) inhibitor aspirin, the P2Y12 antagonist clopidogrel, and the GPIIb-GPIIIa antagonists.1 However, there remains a significant incidence of arterial thrombosis in patients treated with currently available antiplatelet therapy. Novel P2Y12 antagonists such as the recently FDA-approved prasugrel, along with ticagrelor, cangrelor, and elinogrel, have advantages over clopidogrel, including more rapid, less variable, and more complete inhibition of platelet function. Currently ongoing phase 3 studies will determine whether these new P2Y12 antagonists will result in better and/or more rapid antithrombotic effects than clopidogrel, without an unacceptable increase in hemorrhagic or other side effects, as has been recently reported in some clinical settings for prasugrel16,18,19 and ticagrelor.25 PAR1 antagonists are also undergoing phase 3 trials,38 and many other novel antiplatelet agents are under investigation as antithrombotic agents.
An improved understanding of the mechanisms by which platelets become activated has been essential to the development of novel and improved antiplatelet therapies. A fundamental question is whether improved prevention and treatment of platelet-dependent thrombosis can be separated from a resultant increase in hemorrhagic side effects. The ultimate goal of drug discovery in the field of antiplatelet therapy for the treatment of cardiovascular disease is to find an agent that is effective in preventing thrombosis without causing any increase in bleeding or other side effects.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Lilly/Daiichi Sankyo, Takeda, and GLSynthesis, and has served on a data-monitoring committee for a clinical trial for Lilly/Daiichi Sankyo. Off-label drug use: None disclosed.
Correspondence
Alan D. Michelson, MD, Director, Center for Platelet Research Studies, Children's Hospital Boston, Karp 08213, 300 Longwood Ave, Boston, MA 02115-5737; Phone: (617) 919-2116; Fax: (617) 730-4631; e-mail: alan.michelson@childrens.harvard.edu.
